อ่าน 9 นาที
ฟราแท็กซิน
ฟราแท็กซิน เป็นโปรตีน ที่ ในมนุษย์ถูกเข้ารหัสโดย ยีน FXN [ 5 ] [ 6 ]
ฟราแท็กซิน
| เอฟเอ็กซ์เอ็น | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| ตัวระบุ | |||||||||||||||||||||||||||||||||||||||||||||||||||
| ชื่อเรียกอื่น | FXN , CyaY, FA, FARR, FRDA, X25, ฟราแทกซิน | ||||||||||||||||||||||||||||||||||||||||||||||||||
| รหัสภายนอก | โอมิม : 606829 ; เอ็มจีไอ : 1096879 ; โฮโมโลยีน : 47908 ; GeneCards : FXN ; OMA : FXN - ออโธโลจี | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| วิกิดาต้า | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||





ฟราแท็กซินเป็นโปรตีนที่ในมนุษย์ถูกเข้ารหัสโดยยีนFXN [ 5 ] [ 6 ]
โปรตีนฟราแท็กซิน (Frataxin) ตั้งอยู่ในไมโทคอนเดรีย และ mRNA ของฟราแท็กซินมักแสดงออกในเนื้อเยื่อที่มีอัตราการเผาผลาญสูง หน้าที่ของฟราแท็กซินยังไม่ชัดเจน แต่เชื่อว่ามีส่วนเกี่ยวข้องกับการประกอบกลุ่มเหล็ก-กำมะถัน มีการเสนอว่ามันอาจทำหน้าที่เป็นทั้งโปรตีนช่วยนำพาธาตุเหล็ก (iron chaperone) หรือโปรตีนเก็บสะสมธาตุเหล็ก การแสดงออกของฟราแท็กซินที่ลดลงเป็นสาเหตุของโรคอะแท็กเซียของฟรีดไรช์ (Friedreich's ataxia )
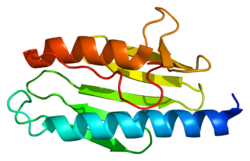
โครงสร้าง
การวิเคราะห์โครงสร้างผลึกด้วยรังสีเอกซ์แสดงให้เห็นว่าฟราแท็กซินของมนุษย์ประกอบด้วยแผ่นเบต้า ที่รองรับ เกลียวอัลฟาคู่ขนานทำให้เกิดโครงสร้างแซนด์วิชอัลฟาเบต้าที่กะทัดรัด[ 7 ] ฟราแท็กซินโฮโมล็อกในสปีชีส์อื่น ๆ มีลักษณะคล้ายกัน โดยมีโครงสร้างหลักเดียวกัน อย่างไรก็ตาม ลำดับหางของฟราแท็กซินที่ยื่นออกมาจากปลายเกลียวหนึ่งนั้นแตกต่างกันในลำดับและความยาว ฟราแท็กซินของมนุษย์มีลำดับหางที่ยาวกว่าฟราแท็กซินที่พบในแบคทีเรียหรือยีสต์ มีการตั้งสมมติฐานว่าจุดประสงค์ของหางคือการทำให้โปรตีนมีเสถียรภาพ[ 7 ]
เช่นเดียวกับ โปรตีนไมโตคอนเดรียส่วนใหญ่ฟราแท็กซินถูกสังเคราะห์ในไรโบโซมไซโตพลาสมิ ก เป็นโมเลกุลตั้งต้นขนาดใหญ่ที่มีลำดับเป้าหมายไมโตคอนเดรีย เมื่อเข้าสู่ไมโตคอนเดรีย โมเลกุลจะถูกย่อยสลายด้วย ปฏิกิริยา โปรตีโอไลติกเพื่อให้ได้ฟราแท็กซินที่สมบูรณ์[ 8 ]
การทำงาน
ฟราแท็กซินมีตำแหน่งอยู่ที่ไมโทคอนเดรีย หน้าที่ของฟราแท็กซินยังไม่ชัดเจนนัก แต่ดูเหมือนว่าจะเกี่ยวข้องกับการประกอบคลัสเตอร์เหล็ก-ซัลเฟอร์มีการเสนอว่ามันทำหน้าที่เป็นทั้งตัวช่วยนำ เหล็ก หรือโปรตีนเก็บสะสมเหล็ก[ 9 ]
mRNAของ Frataxin แสดงออกอย่างเด่นชัดในเนื้อเยื่อ ที่มี อัตราการเผาผลาญ สูง(รวมถึงตับ ไต ไขมันสีน้ำตาล และหัวใจ) โฮโมล็อกของ Frataxin ในหนูและยีสต์มีลำดับเป้าหมายไมโทคอนเดรียที่ปลาย N ที่เป็นไปได้ และ พบว่า Frataxin ของมนุษย์มีการอยู่ร่วมกันกับโปรตีนไมโทคอนเดรีย นอกจากนี้ การหยุดชะงักของยีนยีสต์ยังแสดงให้เห็นว่าส่งผลให้เกิดความผิดปกติของไมโทคอนเด รีย ดังนั้น โรค Friedreich's ataxiaจึงเชื่อว่าเป็นโรค ไมโทคอนเดรีย ที่เกิดจากการกลายพันธุ์ในจีโนมนิวเคลียร์ (โดยเฉพาะการขยายตัวของลำดับ GAA สามตัวซ้ำในอินทรอนิกส์ของยีน FXN ซึ่งเข้ารหัสโปรตีน Frataxin) [ 5 ] [ 10 ] [ 11 ]
ความสำคัญทางคลินิก
การแสดงออกของฟราแท็กซินที่ลดลงเป็นสาเหตุของ โรค อะแท็กเซียของฟรีดไรช์ (FRDA) ซึ่งเป็น โรคความเสื่อมของ ระบบประสาทการลดลงของการแสดงออกของยีนฟราแท็กซินอาจเกิดจากการปิดกั้นการถอดรหัสของยีนฟราแท็กซินเนื่องจาก การดัดแปลง เอพิเจเนติกในหน่วยโครโมโซม[ 12 ]หรือจากความไม่สามารถในการตัดต่อซ้ำ GAA ที่ขยายตัวในอินทรอน แรก ของพรี-mRNA ดังที่พบในแบคทีเรีย[ 13 ]และเซลล์มนุษย์[ 14 ]หรือทั้งสองอย่าง การขยายตัวของลำดับนิวคลีโอไทด์สามตัวซ้ำ GAA ในอินทรอนส่งผลให้เกิดโรคอะแท็กเซียของฟรีดไรช์[ 15 ] ลำดับซ้ำที่ขยายตัวนี้ทำให้เกิด การก่อตัวของ R-loop และการใช้ ออลิโกนิวคลีโอไทด์ที่กำหนดเป้าหมายซ้ำเพื่อทำลาย R-loop สามารถกระตุ้นการแสดงออกของฟราแท็กซินได้อีกครั้ง[ 16 ]
ผู้ป่วย FRDA ร้อยละ 96 มีการขยายตัวของลำดับนิวคลีโอไทด์ GAA ซ้ำในอินทรอน 1 ของอัลลีล ทั้งสอง ของยีนFXN [ 17 ] โดยรวมแล้ว สิ่งนี้ทำให้ การสังเคราะห์ mRNA ของฟราแท็กซินลดลง และโปรตีนฟราแท็กซินลดลง (แต่ไม่ใช่หายไป) ในผู้ป่วย FRDA (ผู้ป่วย FRDA บางกลุ่มมีการขยายตัวของ GAA ในโครโมโซมหนึ่ง และมีการกลายพันธุ์แบบจุดใน เอ็กซอน FXNในโครโมโซมอีกข้างหนึ่ง) ในกรณีทั่วไป ความยาวของอัลลีลที่มีการขยายตัวของ GAA ที่สั้นกว่าจะสัมพันธ์ผกผันกับระดับฟราแท็กซิน เนื้อเยื่อส่วนปลายของผู้ป่วย FRDA มักจะมีระดับฟราแท็กซินน้อยกว่าร้อยละ 10 ของระดับที่พบในผู้ที่ไม่ได้รับ ผลกระทบ [ 17 ] ระดับฟราแท็กซินที่ต่ำลงส่งผลให้โรคเริ่มแสดงอาการเร็วขึ้นและดำเนินไปอย่างรวดเร็ว
FRDA มีลักษณะเฉพาะคืออาการเดินเซ การสูญเสียการรับรู้ และโรคกล้ามเนื้อหัวใจ สาเหตุที่การขาดฟราแท็กซินทำให้เกิดอาการเหล่านี้ยังไม่ชัดเจนนัก ในระดับเซลล์นั้น เกี่ยวข้องกับการสะสมของธาตุเหล็กในไมโทคอนเดรียและความไวต่อสารออกซิแดนต์ที่เพิ่มขึ้น ด้วยเหตุผลที่ยังไม่เป็นที่เข้าใจดีนัก สิ่งนี้ส่งผลกระทบต่อเนื้อเยื่อของปมประสาทรากหลัง สมองน้อยและกล้ามเนื้อหัวใจ เป็นหลัก [ 8 ]
การศึกษาในสัตว์
ในหนู การปิดใช้งาน โฮโมล็อก FXN ( Frda ) อย่างสมบูรณ์นั้นเป็นอันตรายถึงชีวิตในระยะตัวอ่อนตอนต้น[ 18 ] แม้ว่าสิ่งมีชีวิตเกือบทั้งหมดจะแสดงโฮโมล็อกฟราแท็กซิน แต่การทำซ้ำ GAA ในอินทรอน 1 มีอยู่เฉพาะในมนุษย์และไพรเมต อื่นๆ เท่านั้น ดังนั้นการกลายพันธุ์ที่ทำให้เกิด FDRA จึงไม่สามารถเกิดขึ้นตามธรรมชาติในสัตว์อื่นๆ ได้ นักวิทยาศาสตร์ได้พัฒนาทางเลือกหลายอย่างเพื่อสร้างแบบจำลองโรคนี้ในหนู วิธีหนึ่งคือการปิดการแสดงออกของฟราแท็กซินในเนื้อเยื่อประเภทใดประเภทหนึ่งโดยเฉพาะ เช่น หัวใจ (หนูที่ได้รับการดัดแปลงด้วยวิธีนี้เรียกว่า MCK) เซลล์ประสาททั้งหมด (NSE) หรือเฉพาะไขสันหลังและสมองน้อย (PRP) [ 19 ] อีกวิธีหนึ่งเกี่ยวข้องกับการแทรกการขยายตัวของ GAA เข้าไปในอินทรอนแรกของ ยีน FXN ของหนู ซึ่งควรจะยับยั้งการผลิตฟราแท็กซิน เช่นเดียวกับในมนุษย์ หนูที่เป็นโฮโมไซกัสสำหรับยีนที่ดัดแปลงนี้เรียกว่า KIKI (knock-in knock-in) และเฮเทอโรไซกัสแบบผสมที่เกิดจากการผสมพันธุ์หนู KIKI กับหนูที่ยีน frataxin ถูกน็อคเอาท์เรียกว่า KIKO (knock-in knock-out) อย่างไรก็ตาม แม้แต่หนู KIKO ก็ยังคงแสดงระดับ frataxin ปกติอยู่ที่ 25-36% และแสดงอาการเพียงเล็กน้อย วิธีการสุดท้ายเกี่ยวข้องกับการสร้าง หนู ทรานส์เจนิกที่มียีน frataxin ของมนุษย์เวอร์ชันที่ขยาย GAA หนูเหล่านี้เรียกว่า YG22R (ลำดับ GAA หนึ่งลำดับที่มีการทำซ้ำ 190 ครั้ง) และ YG8R (ลำดับ GAA สองลำดับที่มีการทำซ้ำ 90 และ 190 ครั้ง) หนูเหล่านี้แสดงอาการคล้ายกับผู้ป่วยที่เป็นมนุษย์[ 19 ]
การแสดงออกของฟราแท็กซินมากเกินไปในDrosophilaแสดงให้เห็นถึงการเพิ่มขึ้นของความสามารถในการต้านอนุมูลอิสระ ความต้านทานต่อการทำร้ายจากความเครียดออกซิเดชัน และอายุยืนยาว[ 20 ]ซึ่งสนับสนุนทฤษฎีที่ว่าบทบาทของฟราแท็กซินคือการปกป้องไมโตคอนเดรียจากความเครียดออกซิเดชันและความเสียหายของเซลล์ที่ตามมา
ไฟโบรบลาสต์จากหนูทดลองที่เป็นแบบจำลองของ FRDA และไฟโบรบลาสต์ของผู้ป่วย FRDA แสดงให้เห็นระดับการแตกของดีเอ็นเอแบบสองสาย ที่เพิ่มขึ้น [ 21 ]ระบบนำส่งยีนเลนติไวรัสถูกใช้เพื่อนำส่งยีนฟราแท็กซินไปยังหนูทดลองที่เป็นแบบจำลองของ FRDA และเซลล์ของผู้ป่วย และส่งผลให้มีการฟื้นฟูการแสดงออกของmRNA ฟราแท็กซิน และโปรตีนฟราแท็กซินในระยะยาว การฟื้นฟูการแสดงออกของยีนฟราแท็กซินนี้มาพร้อมกับการลดลงอย่างมากของจำนวนการแตกของดีเอ็นเอแบบสองสาย[ 21 ]ฟราแท็กซินที่บกพร่องในเซลล์ FRDA ดูเหมือนจะทำให้ความสามารถในการซ่อมแซมความเสียหายของดีเอ็นเอ ลดลง และอาจมีส่วนทำให้เกิดการเสื่อมของระบบประสาท[ 21 ]
ปฏิสัมพันธ์
พบว่าฟราแท็กซินมีปฏิสัมพันธ์ ทางชีวภาพ กับเอนไซม์PMPCB [ 22 ]
อ่านเพิ่มเติม
- Thierbach R, Drewes G, Fusser M, Voigt A, Kuhlow D, Blume U, Schulz TJ, Reiche C, Glatt H, Epe B, Steinberg P, Ristow M (พ.ย. 2010). "โปรตีนฟราแท็กซินของโรคอะแท็กเซียของฟรีดไรช์ปรับเปลี่ยนการซ่อมแซมการตัดฐานดีเอ็นเอในโปรคาริโอตและสัตว์เลี้ยงลูกด้วยนม"วารสารชีวเคมี 432 ( 1): 165– 72. doi : 10.1042/BJ20101116 . PMC 2976068 . PMID 20819074 .
- มอนแตร์มินี แอล, โรดิอุส เอฟ, เปียเนเซ่ แอล, โมลโต้ เอ็มดี, คอสเซ่ เอ็ม, กัมปูซาโน่ วี, คาวาลกันติ เอฟ, มอนติเชลลี เอ, ปาเลา เอฟ, กยาปาย จี (พ.ย. 1995) "บริเวณวิกฤต ataxia ของฟรีดริชครอบคลุมช่วง 150-kb บนโครโมโซม 9q13 " วารสารอเมริกันพันธุศาสตร์มนุษย์ . 57 (5): 1061–7 . PMC 1801369 . PMID 7485155 .
- Bidichandani SI, Ashizawa T, Patel PI (พฤษภาคม 1997). "โรค Friedreich ataxia ที่ผิดปกติซึ่งเกิดจากภาวะเฮเทอโรไซโกตแบบผสมสำหรับการกลายพันธุ์แบบมิสเซนส์ใหม่และการขยายตัวของไตรเพล็ต GAA"วารสารพันธุศาสตร์มนุษย์อเมริกัน 60 ( 5): 1251– 6. PMC 1712428 . PMID 9150176 .
- แบ็บค็อก เอ็ม, เด ซิลวา ดี, โอ๊คส์ อาร์, เดวิส-แคปแลน เอส, จิราเลอร์สปอง เอส, มอนแตร์มินี แอล, แพนดอลโฟ เอ็ม, แคปแลน เจ (มิ.ย. 1997) "การควบคุมการสะสมธาตุเหล็กในไมโตคอนเดรียโดย Yfh1p ซึ่งเป็นสมมุติฐานที่คล้ายคลึงกันของ Frataxin" ศาสตร์ . 276 (5319): 1709– 12. ดอย : 10.1126/science.276.5319.1709 . PMID9180083 .
- Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (สิงหาคม 1997). "การศึกษาโฮโมล็อกของมนุษย์ หนู และยีสต์บ่งชี้ถึงหน้าที่ของฟราแท็กซินในไมโทคอนเดรีย" Nature Genetics . 16 (4): 345– 51. doi : 10.1038/ng0897-345 . PMID 9241270 . S2CID 5883249 .
- Wilson RB, Roof DM (สิงหาคม 1997). "ภาวะขาดออกซิเจนเนื่องจากการสูญเสียดีเอ็นเอไมโทคอนเดรียในยีสต์ที่ขาดโฮโมล็อกของฟราแท็กซิน" Nature Genetics . 16 (4): 352– 7. doi : 10.1038/ng0897-352 . PMID 9241271 . S2CID 22652291 .
- กัมปูซาโน่ วี, มอนแตร์มินี แอล, ลุตซ์ วาย, โควา แอล, ฮินเดแลง ซี, จิราเลอร์สปอง เอส, ทรอตติเยร์ วาย, คิช เอสเจ, โฟชัวซ์ บี, ทรอยยาส พี, Authier FJ, ดูร์ร์ เอ, มานเดล เจแอล, เวสโควี เอ, แพนดอลโฟ เอ็ม, โคนิก เอ็ม (ต.ค. 1997) "Frataxin จะลดลงในผู้ป่วยที่ Friedreich ataxia และมีความสัมพันธ์กับเยื่อหุ้มไมโตคอนเดรีย " อณูพันธุศาสตร์มนุษย์ . 6 (11): 1771– 80. ดอย : 10.1093/hmg/6.11.1771 . PMID9302253 .
- Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (ต.ค. 1997). "การขาดเอนไซม์อะโคนิเทสและโปรตีนเหล็ก-กำมะถันในไมโทคอนเดรียในโรคอะแท็กเซียของฟรีดไรช์" Nature Genetics . 17 (2): 215– 7. doi : 10.1038/ng1097-215 . PMID 9326946 . S2CID 23151137 .
- Jiralerspong S, Liu Y, Montermini L, Stifani S, Pandolfo M (1997). "Frataxin แสดงการแสดงออกเฉพาะเนื้อเยื่อที่ควบคุมตามพัฒนาการในตัวอ่อนของหนู" Neurobiology of Disease . 4 (2): 103– 13. doi : 10.1006/nbdi.1997.0139 . PMID 9331900 . S2CID 6520439 .
- Koutnikova H, Campuzano V, Koenig M (ก.ย. 1998). "การเจริญเติบโตของฟราแท็กซินชนิดปกติและชนิดกลายพันธุ์โดยเปปติเดสประมวลผลไมโทคอนเดรีย"พันธุศาสตร์โมเลกุลของมนุษย์7 (9): 1485– 9. doi : 10.1093/hmg/7.9.1485 . PMID 9700204 .
- Zühlke C, Laccone F, Cossée M, Kohlschütter A, Koenig M, Schwinger E (กรกฎาคม 1998). "การกลายพันธุ์ของรหัสเริ่มต้นในยีน FRDA1: การวิเคราะห์การเชื่อมโยงของสามตระกูลที่มีการเปลี่ยน ATG เป็น ATT ชี้ให้เห็นถึงบรรพบุรุษร่วมกันเพียงหนึ่งเดียว" Human Genetics . 103 (1): 102– 5. doi : 10.1007/s004390050791 . PMID 9737785 . S2CID 26999143 .
- Bartolo C, Mendell JR, Prior TW (ต.ค. 1998). "การระบุการกลายพันธุ์แบบมิสเซนส์ในผู้ป่วยโรคอะแท็กเซียของฟรีดไรช์: ผลกระทบต่อการวินิจฉัยและการศึกษาผู้พาหะ" American Journal of Medical Genetics . 79 (5): 396– 9. doi : 10.1002/(SICI)1096-8628(19981012)79:5<396::AID-AJMG13>3.0.CO;2-M . PMID 9779809 .
- Cossée M, Dürr A, Schmitt M, Dahl N, Trouillas P, Allinson P, Kostrzewa M, Nivelon-Chevallier A, Gustavson KH, Kohlschütter A, Müller U, Mandel JL, Brice A, Koenig M, Cavalcanti F, Tammaro A, De Michele G, Filla A, Cocozza S, Labuda M, มอนแตร์มินี แอล, Poirier J, Pandolfo M (ก.พ. 1999) การสูญเสียของฟรีดริช: การกลายพันธุ์แบบจุดและการนำเสนอทางคลินิกของสารประกอบเฮเทอโรไซโกตพงศาวดารของประสาทวิทยา . 45 (2): 200– 6. ดอย : 10.1002/1531-8249(199902)45:2<200::AID-ANA10>3.0.CO;2-U . PMID 9989622 . S2CID 24885238 .
- Coppola G, De Michele G, Cavalcanti F, Pianese L, Perretti A, Santoro L, Vita G, Toscano A, Amboni M, Grimaldi G, Salvatore E, Caruso G, Filla A (พฤษภาคม 1999). "ทำไมผู้ป่วยโรค Friedreich's ataxia บางรายจึงยังคงมีปฏิกิริยาตอบสนองของเอ็น? การศึกษาทางคลินิก ระบบประสาท และโมเลกุล" วารสารประสาทวิทยา 246 ( 5): 353– 7. doi : 10.1007/s004150050362 . PMID 10399865 . S2CID 7367457 .
- Branda SS, Cavadini P, Adamec J, Kalousek F, Taroni F, Isaya G (สิงหาคม 1999). "ฟราแท็กซินของยีสต์และมนุษย์ถูกประมวลผลให้เป็นรูปแบบที่สมบูรณ์ในสองขั้นตอนต่อเนื่องกันโดยเปปติเดสประมวลผลไมโทคอนเดรีย"วารสารเคมีชีวภาพ 274 ( 32): 22763– 9. doi : 10.1074/jbc.274.32.22763 . PMID 10428860 .
- Gordon DM, Shi Q, Dancis A, Pain D (พ.ย. 1999). "การเจริญเติบโตของฟราแท็กซินภายในไมโทคอนเดรียของสัตว์เลี้ยงลูกด้วยนมและยีสต์: การประมวลผลขั้นตอนเดียวโดยเมทริกซ์โปรเซสซิ่งเปปติเดส" Human Molecular Genetics . 8 (12): 2255– 62. doi : 10.1093/hmg/8.12.2255 . PMID 10545606 .
- Forrest SM, Knight M, Delatycki MB, Paris D, Williamson R, King J, Yeung L, Nassif N, Nicholson GA (สิงหาคม 1998). "ความสัมพันธ์ของลักษณะทางคลินิกในโรค Friedreich ataxia กับตำแหน่งของการกลายพันธุ์แบบจุดในยีน FRDA" Neurogenetics . 1 (4): 253– 7. doi : 10.1007/s100480050037 . PMID 10732799 . S2CID 7463903 .
- Al-Mahdawi S, Pook M, Chamberlain S (กรกฎาคม 2543). "การกลายพันธุ์แบบมิสเซนส์ชนิดใหม่ (L198R) ในยีน Friedreich's ataxia" . Human Mutation . 16 (1): 95. doi : 10.1002/1098-1004(200007)16:1<95::AID-HUMU29>3.0.CO;2-E . PMID 10874325 . S2CID 26295274 .
ลิงก์ภายนอก
- รายการ GeneReviews/NCBI/NIH/UW บน Friedreich Ataxia
- frataxin ในฐานข้อมูล Medical Subject Headings (MeSH) ของหอสมุดแห่งชาติสหรัฐอเมริกา
- ภาพรวมของข้อมูลโครงสร้างทั้งหมดที่มีอยู่ในPDBสำหรับUniProt : Q16595 (Frataxin, mitochondrial) ที่PDBe- KB
สรุปเนื้อหา
ข้อมูลสำคัญจากบทความ
ข้อมูลสำคัญเกี่ยวกับ ฟราแท็กซิน
ฟราแท็กซิน เป็นโปรตีน ที่ ในมนุษย์ถูกเข้ารหัสโดย ยีน FXN [ 5 ] [ 6 ]
โครงสร้าง
การวิเคราะห์โครงสร้างผลึกด้วยรังสีเอกซ์ แสดงให้เห็นว่าฟราแท็กซินของมนุษย์ประกอบด้วย แผ่นเบต้า ที่รองรับ เกลียวอัลฟา คู่ขนานทำให้เกิดโครงสร้างแซนด์วิชอัลฟาเบต้าที่กะทัดรัด [ 7 ] ฟราแท็ กซินโฮโมล็อก ในสปีชีส์อื่น ๆ มีลักษณะคล้ายกัน โดยมีโครงสร้างหลักเดียวกัน...
การทำงาน
ฟราแท็กซินมีตำแหน่งอยู่ที่ ไมโทคอนเดรี ย หน้าที่ของฟราแท็กซินยังไม่ชัดเจนนัก แต่ดูเหมือนว่าจะเกี่ยวข้องกับการประกอบ คลัสเตอร์เหล็ก-ซัลเฟอร์ มีการเสนอว่ามันทำหน้าที่เป็นทั้งตัว ช่วยนำ เหล็ก หรือโปรตีนเก็บสะสมเหล็ก [ 9 ]
ความสำคัญทางคลินิก
การแสดงออกของฟราแท็กซินที่ลดลงเป็นสาเหตุของ โรค อะแท็กเซียของฟรีดไรช์ (FRDA) ซึ่งเป็น โรคความเสื่อมของ ระบบประสาท การลดลงของการแสดงออกของยีนฟราแท็กซินอาจเกิดจากการปิดกั้นการถอดรหัสของยีนฟราแท็กซินเนื่องจาก การดัดแปลง เอพิเจเนติก ในหน่วยโครโมโซม [ 12 ]...