การเชื่อมต่อ (ระดับโมเลกุล)
| คำศัพท์เกี่ยวกับการเชื่อมต่อ |
|---|
|
ในสาขาการสร้างแบบจำลองโมเลกุลการด็อกกิ้งเป็นวิธีการที่ทำนายทิศทางที่ต้องการของโมเลกุลหนึ่งกับโมเลกุลที่สองเมื่อลิแกนด์และเป้าหมายจับกันเพื่อสร้างคอมเพล็กซ์ ที่ เสถียร[ 1 ]ความรู้เกี่ยวกับทิศทางที่ต้องการนั้นอาจนำไปใช้ในการทำนายความแข็งแรงของการเชื่อมโยงหรือความสัมพันธ์ในการจับกันระหว่างโมเลกุลสองโมเลกุลโดยใช้ฟังก์ชันการให้คะแนนเป็นต้น

การจับคู่กันระหว่างโมเลกุลที่มีความสำคัญทางชีววิทยา เช่นโปรตีนเปปไทด์กรดนิวคลีอิกคาร์โบไฮเดรตและไขมันมีบทบาทสำคัญในการส่งสัญญาณนอกจากนี้ การวางตัวสัมพัทธ์ของคู่พันธะอาจส่งผลต่อประเภทของสัญญาณที่เกิดขึ้น (เช่นการกระตุ้นหรือการยับยั้ง ) ดังนั้น การจับคู่จึงมีประโยชน์ในการทำนายทั้งความแรงและประเภทของสัญญาณที่เกิดขึ้น
การจับคู่โมเลกุลเป็นหนึ่งในวิธีการที่ใช้บ่อยที่สุดในการออกแบบยาโดยอาศัยโครงสร้างเนื่องจากมีความสามารถในการทำนายโครงสร้างการจับของ ลิแกนด์ โมเลกุลขนาดเล็กกับตำแหน่งการจับ เป้าหมายที่เหมาะสม การกำหนดลักษณะพฤติกรรมการจับมีบทบาทสำคัญในการออกแบบยาอย่างมีเหตุผลรวมถึงการอธิบายกระบวนการทางชีวเคมีพื้นฐาน[ 2 ] [ 3 ]ดังนั้น การจับคู่จึงมีประโยชน์ในการค้นหาลิแกนด์ใหม่สำหรับเป้าหมายโดยการคัดกรองไลบรารีสารประกอบเสมือนขนาดใหญ่ และเป็นจุดเริ่มต้นสำหรับการเพิ่มประสิทธิภาพลิแกนด์หรือการตรวจสอบกลไกการออกฤทธิ์[ 4 ]
นิยามของปัญหา
เราอาจมองว่าการจับคู่โมเลกุลเป็นปัญหาแบบ"กุญแจและแม่กุญแจ"ซึ่งเราต้องการหาทิศทางสัมพัทธ์ที่ถูกต้องของ"กุญแจ"ที่จะเปิด"แม่กุญแจ" (เช่น รูกุญแจอยู่ตรงไหนบนพื้นผิวของแม่กุญแจ ต้องหมุนกุญแจไปในทิศทางใดหลังจากเสียบเข้าไปแล้ว เป็นต้น) ในที่นี้ โปรตีนอาจเปรียบได้กับ "แม่กุญแจ" และลิแกนด์อาจเปรียบได้กับ "กุญแจ" การจับคู่โมเลกุลอาจนิยามได้ว่าเป็นปัญหาการหาค่าเหมาะสมที่สุด ซึ่งจะอธิบายถึงทิศทาง "เหมาะสมที่สุด" ของลิแกนด์ที่จับกับโปรตีนเป้าหมาย อย่างไรก็ตาม เนื่องจากทั้งลิแกนด์และโปรตีนมีความยืดหยุ่น การเปรียบเทียบแบบ "มือกับถุงมือ"จึงเหมาะสมกว่า"กุญแจและแม่กุญแจ" [ 5 ] ในระหว่างกระบวนการจับคู่ ลิแกนด์และโปรตีนจะปรับโครงสร้างเพื่อให้ได้ "ความเหมาะสมที่สุด" โดยรวม และการปรับโครงสร้างแบบนี้ที่ส่งผลให้เกิดการจับกันโดยรวมเรียกว่า" ความเหมาะสมที่เหนี่ยวนำ" [ 6 ]
งานวิจัยด้านการจับคู่โมเลกุล (Molecular docking) มุ่งเน้นไปที่การจำลอง กระบวนการ จดจำโมเลกุล ด้วยวิธีการคำนวณ โดยมี เป้าหมายเพื่อหาโครงสร้างที่เหมาะสมที่สุดสำหรับทั้งโปรตีนและลิแกนด์ รวมถึงการวางตัวสัมพัทธ์ระหว่างโปรตีนและลิแกนด์ เพื่อให้พลังงานอิสระของระบบโดยรวมมีค่าน้อยที่สุด
วิธีการเชื่อมต่อ
มีสองแนวทางที่ได้รับความนิยมเป็นพิเศษในกลุ่มผู้เชี่ยวชาญด้านการจับคู่โมเลกุล
- แนวทางหนึ่งใช้เทคนิคการจับคู่ที่อธิบายโปรตีนและลิแกนด์ว่าเป็นพื้นผิวที่เสริมกัน[ 7 ] [ 8 ] [ 9 ]
- แนวทางที่สองจำลองกระบวนการด็อกกิ้งจริงซึ่งพลังงานปฏิสัมพันธ์แบบคู่ระหว่างลิแกนด์กับโปรตีนจะถูกคำนวณ[ 10 ]
ทั้งสองแนวทางต่างก็มีข้อดีและข้อจำกัดที่สำคัญ ซึ่งจะกล่าวถึงต่อไปนี้
ความสมบูรณ์ของรูปทรง
วิธีการจับคู่ทางเรขาคณิต/ความเสริมกันของรูปร่างอธิบายโปรตีนและลิแกนด์เป็นชุดของคุณลักษณะที่ทำให้สามารถด็อกกิ้งได้[ 11 ]คุณลักษณะเหล่านี้อาจรวมถึง ตัวอธิบาย พื้นผิวโมเลกุล / พื้นผิวเสริมกันในกรณีนี้ พื้นผิวโมเลกุลของตัวรับจะถูกอธิบายในแง่ของพื้นที่ผิวที่เข้าถึงตัวทำละลายได้และพื้นผิวโมเลกุลของลิแกนด์จะถูกอธิบายในแง่ของคำอธิบายพื้นผิวที่ตรงกัน ความเสริมกันระหว่างสองพื้นผิวเท่ากับคำอธิบายการจับคู่รูปร่างที่อาจช่วยในการค้นหาตำแหน่งเสริมกันของการด็อกกิ้งโมเลกุลเป้าหมายและลิแกนด์ อีกแนวทางหนึ่งคือการอธิบายคุณลักษณะที่ไม่ชอบน้ำของโปรตีนโดยใช้การโค้งงอในอะตอมของสายโซ่หลัก อีกแนวทางหนึ่งคือการใช้เทคนิคตัวอธิบายรูปร่างฟูริเยร์[ 12 ] [ 13 ] [ 14 ]ในขณะที่วิธีการที่ใช้ความเสริมกันของรูปร่างมักจะรวดเร็วและแข็งแกร่ง แต่โดยทั่วไปแล้วไม่สามารถจำลองการเคลื่อนไหวหรือการเปลี่ยนแปลงแบบไดนามิกในโครงสร้างของลิแกนด์/โปรตีนได้อย่างแม่นยำ แม้ว่าการพัฒนาล่าสุดจะช่วยให้วิธีการเหล่านี้สามารถตรวจสอบความยืดหยุ่นของลิแกนด์ได้ วิธีการจับคู่รูปร่างสามารถสแกนลิแกนด์หลายพันตัวได้อย่างรวดเร็วภายในเวลาไม่กี่วินาที และสามารถตรวจสอบได้ว่าลิแกนด์เหล่านั้นสามารถจับกับตำแหน่งออกฤทธิ์ ของโปรตีนได้หรือไม่ และโดยทั่วไปแล้วยังสามารถปรับขนาดให้ใช้ได้กับปฏิกิริยาระหว่างโปรตีนกับโปรตีนอีกด้วย นอกจากนี้ยังเหมาะสมกับวิธีการที่ใช้ ฟาร์มาโคฟอร์มากกว่าเนื่องจากใช้คำอธิบายทางเรขาคณิตของลิแกนด์เพื่อค้นหาการจับที่เหมาะสมที่สุด
การจำลอง
การจำลองกระบวนการด็อกกิ้งนั้นซับซ้อนกว่ามาก ในวิธีการนี้ โปรตีนและลิแกนด์จะอยู่ห่างกันในระยะทางกายภาพ และลิแกนด์จะหาตำแหน่งของตัวเองในบริเวณออกฤทธิ์ของโปรตีนหลังจาก "การเคลื่อนที่" จำนวนหนึ่งในพื้นที่โครงสร้างของมัน การเคลื่อนที่เหล่านี้รวมถึงการเปลี่ยนแปลงรูปร่างของวัตถุแข็ง เช่น การเลื่อนและการหมุน รวมถึงการเปลี่ยนแปลงภายในโครงสร้างของลิแกนด์ เช่น การหมุนมุมบิด การเคลื่อนที่แต่ละครั้งในพื้นที่โครงสร้างของลิแกนด์จะทำให้เกิดต้นทุนพลังงานรวมของระบบ ดังนั้น พลังงานรวมของระบบจึงถูกคำนวณหลังจากการเคลื่อนที่แต่ละครั้ง
ข้อได้เปรียบที่เห็นได้ชัดของการจำลองการจับคู่โมเลกุลคือ สามารถรวมความยืดหยุ่นของลิแกนด์ได้อย่างง่ายดาย ในขณะที่เทคนิคการเสริมรูปร่างต้องใช้วิธีการที่ซับซ้อนเพื่อรวมความยืดหยุ่นในลิแกนด์ นอกจากนี้ การจำลองการจับคู่โมเลคูลยังจำลองความเป็นจริงได้แม่นยำกว่า ในขณะที่เทคนิคการเสริมรูปร่างเป็นเพียงนามธรรมมากกว่า
เห็นได้ชัดว่าการจำลองนั้นใช้ทรัพยากรการคำนวณสูง เนื่องจากต้องสำรวจภูมิทัศน์พลังงานขนาดใหญ่ เทคนิคแบบกริด วิธีการปรับให้เหมาะสม และความเร็วของคอมพิวเตอร์ที่เพิ่มขึ้น ทำให้การจำลองการเชื่อมต่อยานอวกาศมีความสมจริงมากขึ้น
กลไกการเชื่อมต่อ
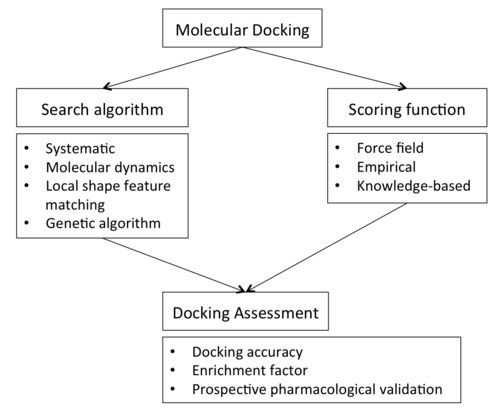
ในการดำเนินการคัดกรองการจับคู่ (docking screen) ข้อกำหนดแรกคือโครงสร้างของโปรตีนที่สนใจ โดยปกติโครงสร้างจะถูกกำหนดโดยใช้เทคนิคทางชีวฟิสิกส์ เช่น
แต่ยังสามารถได้มาจาก การสร้าง แบบจำลองโฮโมโลยีหรือจากการคาดการณ์ของ AlphaFold ได้อีกด้วย [ 15 ]โครงสร้างโปรตีนนี้และฐานข้อมูลของลิแกนด์ที่มีศักยภาพทำหน้าที่เป็นอินพุตให้กับโปรแกรมด็อกกิ้ง ความสำเร็จของโปรแกรมด็อกกิ้งขึ้นอยู่กับสององค์ประกอบ ได้แก่อัลกอริทึมการค้นหาและฟังก์ชันการให้คะแนน
อัลกอริทึมการค้นหา
ในทางทฤษฎี พื้นที่การค้นหาประกอบด้วยทิศทางและโครงสร้าง ที่เป็นไปได้ทั้งหมด ของโปรตีนที่จับคู่กับลิแกนด์ อย่างไรก็ตาม ในทางปฏิบัติ ด้วยทรัพยากรการคำนวณในปัจจุบัน เป็นไปไม่ได้ที่จะสำรวจพื้นที่การค้นหาอย่างละเอียดถี่ถ้วน — ซึ่งจะเกี่ยวข้องกับการแจงนับการบิดเบี้ยวที่เป็นไปได้ทั้งหมดของแต่ละโมเลกุล (โมเลกุลมีความเคลื่อนไหวและมีอยู่ในกลุ่มของสถานะโครงสร้าง) และ ทิศทาง การหมุนและการแปล ที่เป็นไปได้ทั้งหมด ของลิแกนด์เมื่อเทียบกับโปรตีนในระดับความละเอียด ที่กำหนด โปรแกรมด็อกกิ้งส่วนใหญ่ที่ใช้จะคำนึงถึงพื้นที่โครงสร้างทั้งหมดของลิแกนด์ (ลิแกนด์ที่ยืดหยุ่นได้) และหลายโปรแกรมพยายามสร้างแบบจำลองตัวรับโปรตีนที่ยืดหยุ่นได้ แต่ละ "ภาพสแนปช็อต" ของคู่เรียกว่าโพส[ 16 ]
มีการนำกลยุทธ์การค้นหาโครงสร้างที่หลากหลายมาใช้กับลิแกนด์และตัวรับ ซึ่งรวมถึง:
- การค้นหา แรงบิดอย่างเป็นระบบหรือแบบสุ่ม เกี่ยวกับพันธะที่หมุนได้
- การจำลองพลศาสตร์โมเลกุล
- ใช้อัลกอริธึมทางพันธุกรรมเพื่อ "พัฒนา" โครงสร้างที่มีพลังงานต่ำแบบใหม่ โดยคะแนนของแต่ละท่าทางจะทำหน้าที่เป็นฟังก์ชันความเหมาะสมที่ใช้ในการคัดเลือกแต่ละบุคคลสำหรับรอบถัดไป
ความยืดหยุ่นของลิแกนด์
โครงสร้างของลิแกนด์อาจถูกสร้างขึ้นในกรณีที่ไม่มีตัวรับและทำการด็อกกิ้งในภายหลัง[ 17 ]หรือโครงสร้างอาจถูกสร้างขึ้นแบบเรียลไทม์ในกรณีที่มีช่องว่างการจับของตัวรับ[ 18 ]หรือด้วยความยืดหยุ่นในการหมุนอย่างเต็มที่ของทุกมุมไดเฮดรัลโดยใช้การด็อกกิ้งแบบอิงชิ้นส่วน[ 19 ] การประเมินพลังงาน สนามแรงมักใช้เพื่อเลือกโครงสร้างที่สมเหตุสมผลทางพลังงาน[ 20 ]แต่ก็มีการใช้วิธีการตามความรู้ด้วยเช่นกัน[ 21 ]
เปปไทด์เป็นโมเลกุลที่มีความยืดหยุ่นสูงและมีขนาดค่อนข้างใหญ่ ซึ่งทำให้การสร้างแบบจำลองความยืดหยุ่นของเปปไทด์เป็นงานที่ท้าทาย มีการพัฒนาวิธีการหลายวิธีเพื่อให้สามารถสร้างแบบจำลองความยืดหยุ่นของเปปไทด์ได้อย่างมีประสิทธิภาพในระหว่างการเชื่อมต่อโปรตีนกับเปปไทด์[ 22 ]
ความยืดหยุ่นของตัวรับ
ความสามารถในการคำนวณเพิ่มขึ้นอย่างมากในช่วงทศวรรษที่ผ่านมา ทำให้สามารถใช้วิธีการที่ซับซ้อนและต้องใช้การคำนวณมากขึ้นในการออกแบบยาโดยใช้คอมพิวเตอร์ช่วย อย่างไรก็ตาม การจัดการกับความยืดหยุ่นของตัวรับในวิธีการด็อกกิ้งยังคงเป็นปัญหาที่ยุ่งยาก[ 23 ]สาเหตุหลักของความยากลำบากนี้คือจำนวนองศาอิสระจำนวนมากที่ต้องนำมาพิจารณาในการคำนวณประเภทนี้ อย่างไรก็ตาม การละเลยในบางกรณีอาจนำไปสู่ผลลัพธ์การด็อกกิ้งที่ไม่ดีในแง่ของการทำนายตำแหน่งการจับ[ 24 ]
โครงสร้างคงที่หลายโครงสร้างที่กำหนดโดยการทดลองสำหรับโปรตีนเดียวกันในรูปแบบต่างๆ มักถูกนำมาใช้เพื่อจำลองความยืดหยุ่นของตัวรับ[ 25 ]หรือ อาจค้นหา ไลบรารีโรตาเมอร์ของหมู่ข้างเคียงกรดอะมิโนที่ล้อมรอบช่องการจับเพื่อสร้างรูปแบบโปรตีนทางเลือกแต่สมเหตุสมผลทางพลังงาน[ 26 ] [ 27 ]
ฟังก์ชันการให้คะแนน
โปรแกรมการจับคู่โมเลกุลสร้างตำแหน่งการจับของลิแกนด์ที่เป็นไปได้จำนวนมาก ซึ่งบางส่วนสามารถถูกปฏิเสธได้ทันทีเนื่องจากเกิดการชนกับโปรตีน ส่วนที่เหลือจะถูกประเมินโดยใช้ฟังก์ชันการให้คะแนน ซึ่งรับตำแหน่งการจับเป็นอินพุตและส่งคืนค่าตัวเลขที่บ่งบอกถึงความเป็นไปได้ที่ตำแหน่งนั้นจะแสดงถึงปฏิกิริยาการจับที่เหมาะสม และจัดอันดับลิแกนด์หนึ่งเทียบกับลิแกนด์อื่น
ฟังก์ชันการให้คะแนนส่วนใหญ่เป็น ฟิลด์แรงกลศาสตร์โมเลกุล ตามหลักฟิสิกส์ซึ่งประมาณค่าพลังงานของท่าทางภายในบริเวณการจับยึด ส่วนประกอบต่างๆ ที่มีส่วนในการจับยึดสามารถเขียนได้ในรูปสมการบวก:

ส่วนประกอบต่างๆ ประกอบด้วย ผลกระทบของตัวทำละลาย การเปลี่ยนแปลงโครงสร้างของโปรตีนและลิแกนด์ พลังงานอิสระเนื่องจากปฏิกิริยาระหว่างโปรตีนและลิแกนด์ การหมุนภายใน พลังงานการเชื่อมโยงของลิแกนด์และตัวรับเพื่อสร้างคอมเพล็กซ์เดี่ยว และพลังงานอิสระเนื่องจากการเปลี่ยนแปลงในโหมดการสั่น[ 28 ]พลังงานต่ำ (ลบ) บ่งชี้ถึงระบบที่เสถียรและดังนั้นจึงมีแนวโน้มที่จะเกิดปฏิกิริยาการจับกัน
แนวทางทางเลือกใช้ฟังก์ชันการให้คะแนนที่แก้ไขเพื่อรวมข้อจำกัดตามปฏิสัมพันธ์โปรตีน-ลิแกนด์ที่สำคัญที่ทราบ[ 29 ]หรือศักยภาพตามความรู้ที่ได้มาจากปฏิสัมพันธ์ที่สังเกตได้ในฐานข้อมูลขนาดใหญ่ของโครงสร้างโปรตีน-ลิแกนด์ (เช่นProtein Data Bank ) [ 30 ]
มีโครงสร้างจำนวนมากจากผลึกศาสตร์รังสีเอกซ์สำหรับสารประกอบเชิงซ้อนระหว่างโปรตีนและลิแกนด์ที่มีความสัมพันธ์สูง แต่มีจำนวนน้อยกว่าสำหรับลิแกนด์ที่มีความสัมพันธ์ต่ำ เนื่องจากสารประกอบเชิงซ้อนหลังมักมีความเสถียรน้อยกว่าและจึงยากต่อการตกผลึกมากกว่า ฟังก์ชันการให้คะแนนที่ฝึกฝนด้วยข้อมูลนี้สามารถจับคู่ลิแกนด์ที่มีความสัมพันธ์สูงได้อย่างถูกต้อง แต่ก็จะให้โครงสร้างการจับคู่ที่ดูสมเหตุสมผลสำหรับลิแกนด์ที่ไม่จับกับโปรตีนด้วยเช่นกัน ซึ่งทำให้ เกิดผลลัพธ์ ที่ผิดพลาด จำนวนมาก กล่าว คือ ลิแกนด์ที่คาดว่าจะจับกับโปรตีน แต่ในความเป็นจริงแล้วไม่จับกันเมื่อนำมารวมกันในหลอดทดลอง
วิธีหนึ่งในการลดจำนวนผลบวกเท็จคือการคำนวณพลังงานของท่าทางที่มีคะแนนสูงสุดใหม่โดยใช้เทคนิคที่มีความแม่นยำมากขึ้นแต่ต้องใช้การคำนวณที่ซับซ้อนกว่า เช่นวิธีGeneralized BornหรือPoisson-Boltzmann [ 10 ]
การประเมินการเชื่อมต่อ
ความสัมพันธ์ระหว่างการสุ่มตัวอย่างและฟังก์ชันการให้คะแนนส่งผลต่อความสามารถในการด็อกกิ้งในการทำนายตำแหน่งที่เป็นไปได้หรือความสัมพันธ์ในการจับตัวของสารประกอบใหม่ ดังนั้น โดยทั่วไปแล้วจึงจำเป็นต้องมีการประเมินโปรโตคอลการด็อกกิ้ง (เมื่อมีข้อมูลจากการทดลอง) เพื่อกำหนดความสามารถในการทำนาย การประเมินการด็อกกิ้งสามารถทำได้โดยใช้กลยุทธ์ต่างๆ เช่น:
- การคำนวณความแม่นยำในการเชื่อมต่อ (DA)
- ความสัมพันธ์ระหว่างคะแนนการเชื่อมต่อและการตอบสนองเชิงทดลองหรือการกำหนดปัจจัยการเสริมคุณค่า (EF); [ 31 ]
- ระยะห่างระหว่างส่วนที่จับกับไอออนและไอออนในบริเวณออกฤทธิ์
- การปรากฏของแบบจำลองการปรับตัวแบบเหนี่ยวนำ
ความแม่นยำในการเชื่อมต่อ

ความแม่นยำในการด็อกกิ้ง[ 33 ] [ 34 ]แสดงถึงการวัดความสามารถของโปรแกรมด็อกกิ้งในการจำลองตำแหน่งการจับของลิแกนด์ที่สังเกตได้จากการทดลอง[ 35 ] โดยทั่วไปแล้ว ความเหมาะสมจะถูกวัดโดยการคำนวณค่าเบี่ยงเบนมาตรฐานรากกำลังสอง (RMSD) ของอะตอมที่ไม่ใช่ไฮโดรเจนระหว่างตำแหน่งที่ด็อกกิ้งและโครงสร้างจากการทดลอง[ 32 ]
โดยทั่วไปแล้ว ค่า RMSD ที่ต่ำกว่า 2.0 Å ถือเป็นตัวบ่งชี้ถึงแบบจำลองการเชื่อมต่อที่ประสบความสำเร็จ[ 32 ] [ 36 ]อย่างไรก็ตาม เนื่องจากค่า RMSD โดยทั่วไปจะเพิ่มขึ้นตามจำนวนอะตอมหนักในลิแกนด์ การกำหนดค่าตัดที่ผ่อนปรนมากขึ้นอาจเหมาะสมสำหรับโมเลกุลขนาดใหญ่กว่า
ที่สำคัญคือ ไม่ควรใช้ RMSD เพียงอย่างเดียวในการประเมินคุณภาพการด็อกกิ้ง การประเมินที่ครอบคลุมควรพิจารณาถึงความสามารถของโปรแกรมในการจำลองปฏิสัมพันธ์ระหว่างลิแกนด์และตัวรับที่สำคัญ และความน่าจะเป็นของโครงสร้างที่สร้างขึ้น ซึ่งมีความเกี่ยวข้องอย่างยิ่งเมื่อใช้แนวทางการด็อกกิ้งโมเลกุลแบบอิงการเรียนรู้เชิงลึก[ 37 ]
ปัจจัยเสริม
สามารถประเมินหน้าจอการเชื่อมต่อได้โดยพิจารณาจากความสามารถในการเพิ่มความเข้มข้นของลิแกนด์ที่ออกฤทธิ์ที่ทราบแล้วจากฐานข้อมูลขนาดใหญ่ของโมเลกุล " ล่อ " ที่คาดว่าจะไม่จับกัน [ 31 ] [ 38 ] ในแนวทางนี้ ประสิทธิภาพของโปรโตคอลการเชื่อมต่อจะถูกประเมินจากความสามารถในการจัดอันดับสารประกอบที่ออกฤทธิ์ที่ทราบแล้วจำนวนเล็กน้อยภายในเศษส่วนบนสุดของโมเลกุลที่คัดกรอง แม้จะมีโมเลกุลล่อจำนวนมากอยู่ก็ตาม
ปัจจัยเสริมประสิทธิภาพ (EF) เป็นตัววัดปริมาณความสามารถในการตรวจจับเบื้องต้นนี้ โดยมีนิยามดังนี้:

ที่ไหน:
- Nactivex คือจำนวนสารประกอบออกฤทธิ์ที่พบในกลุ่ม x% แรกของรายการจัด
- NX คือจำนวนโมเลกุลที่อยู่ในกลุ่ม x% แรกของรายการจัด
- N คือจำนวนรวมของสารประกอบที่ออกฤทธิ์ในชุดข้อมูล
- N คือจำนวนโมเลกุลทั้งหมดในชุดข้อมูล
ดังนั้น EF จึงเปรียบเทียบสัดส่วนของสารออกฤทธิ์ที่ถูกค้นพบในกลุ่มย่อยที่มีอันดับสูงสุดกับสัดส่วนที่คาดหวังจากการเลือกแบบสุ่ม ค่า EF ที่มากกว่า 1 บ่งชี้ว่ามีการเพิ่มประสิทธิภาพมากกว่าการเลือกแบบสุ่ม
โดยเฉพาะอย่างยิ่งEF วัดการเพิ่มประสิทธิภาพภายใน 1% อันดับแรกของฐานข้อมูลที่จัดอันดับ และมักใช้ในการประเมินประสิทธิภาพการค้นหาเบื้องต้นในการคัดกรองเสมือนจริง ซึ่งการระบุสารประกอบที่ออกฤทธิ์ในอันดับต้น ๆ ของรายการมีความสำคัญเป็นอย่างยิ่ง
นอกจาก EF แล้ว พื้นที่ใต้ เส้นโค้ง ลักษณะการทำงานของผู้รับ (ROC) (AUC) ยังถูกใช้กันอย่างแพร่หลายเพื่อประเมินประสิทธิภาพการคัดกรองในรายการจัดอันดับทั้งหมด[ 38 ]
ในอนาคต
ผลลัพธ์ที่ได้จากการคัดกรองการจับคู่โมเลกุลจะได้รับการตรวจสอบทางเภสัชวิทยา (เช่น การวัดค่า IC ความสัมพันธ์หรือศักยภาพ ) มีเพียงการศึกษาเชิงคาดการณ์เท่านั้นที่ถือเป็นหลักฐานยืนยันความเหมาะสมของเทคนิคสำหรับเป้าหมายเฉพาะ[ 39 ]ในกรณีของตัวรับที่เชื่อมโยงกับโปรตีน G (GPCRs) ซึ่งเป็นเป้าหมายของยาที่วางจำหน่ายในตลาดมากกว่า 30% การจับคู่โมเลกุลนำไปสู่การค้นพบลิแกนด์ GPCR มากกว่า 500 ชนิด[ 40 ] [ 15 ]
การเปรียบเทียบมาตรฐาน
ศักยภาพของโปรแกรมด็อกกิ้งในการจำลองรูปแบบการจับตัวกันตามที่กำหนดโดยการวิเคราะห์ด้วยรังสีเอกซ์สามารถประเมินได้โดยใช้ชุดข้อมูลมาตรฐานสำหรับการด็อกกิ้งหลายชุด
สำหรับโมเลกุลขนาดเล็ก มีชุดข้อมูลมาตรฐานหลายชุดสำหรับการด็อกกิ้งและการคัดกรองเสมือนจริง เช่นAstex Diverse Setซึ่งประกอบด้วยโครงสร้างผลึก X-ray ของโปรตีน-ลิแกนด์คุณภาพสูง[ 41 ] Directory of Useful Decoys (DUD) สำหรับการประเมินประสิทธิภาพการคัดกรองเสมือนจริง[ 31 ] ชุดข้อมูล LEADS -FRAGสำหรับแฟรกเมนต์[ 42 ]และ ชุดข้อมูล LIT-PCBAสำหรับการเรียนรู้ของเครื่องและการคัดกรองเสมือนจริง[ 43 ]
นอกเหนือจากการพัฒนาชุดข้อมูลแล้ว ยังมีการประเมินเปรียบเทียบขนาดใหญ่เกิดขึ้นอีกด้วย การประเมินเปรียบเทียบฟังก์ชันการให้คะแนน (CASF) ให้การเปรียบเทียบเชิงระบบของฟังก์ชันการด็อกกิ้งและการให้คะแนนโดยใช้คอมเพล็กซ์โปรตีน-ลิแกนด์มาตรฐาน[ 44 ] CASF ประเมินประสิทธิภาพในหลายแง่มุม รวมถึงพลังการด็อกกิ้ง (การทำนายท่าทาง) พลังการให้คะแนน (การทำนายความสัมพันธ์ในการจับ) พลังการจัดอันดับ และพลังการคัดกรอง ผลลัพธ์จากการศึกษา CASF เน้นให้เห็นถึงความแปรปรวนอย่างมากในโปรแกรมการด็อกกิ้งและฟังก์ชันการให้คะแนน โดยเน้นว่าไม่มีวิธีใดวิธีหนึ่งที่ทำได้ดีกว่าวิธีอื่นอย่างสม่ำเสมอในทุกเกณฑ์การประเมิน
การประเมินโปรแกรมการเชื่อมต่อเพื่อศักยภาพในการจำลองโหมดการจับเปปไทด์สามารถประเมินได้โดยใช้บทเรียนสำหรับการประเมินประสิทธิภาพของการเชื่อมต่อและการให้คะแนน (LEADS-PEP) [ 45 ]
แอปพลิเคชัน
ปฏิกิริยาการจับกันระหว่างโมเลกุลขนาดเล็ก (ลิแกนด์) กับ โปรตีน เอนไซม์อาจส่งผลให้เอนไซม์ ทำงานหรือ ถูกยับยั้ง หากโปรตีนนั้นเป็นตัวรับ การจับกันของลิแกนด์อาจส่งผลให้เกิด การกระตุ้นหรือการยับยั้ง การด็อกกิ้งมักใช้กันมากที่สุดในด้านการออกแบบยาเนื่องจากยาส่วนใหญ่เป็น โมเลกุล อินทรีย์ ขนาดเล็ก และการด็อกกิ้งสามารถนำไปใช้กับ:
- การระบุเป้าหมาย – การจับคู่โมเลกุลร่วมกับฟังก์ชันการให้คะแนนสามารถใช้เพื่อคัดกรองฐานข้อมูลขนาดใหญ่ของยาที่มีศักยภาพได้อย่างรวดเร็วในคอมพิวเตอร์เพื่อระบุโมเลกุลที่มีแนวโน้มที่จะจับกับโปรตีนเป้าหมายที่สนใจ (ดูการคัดกรองเสมือนจริง ) เภสัชวิทยาแบบย้อนกลับใช้การจับคู่โมเลกุลเพื่อระบุเป้าหมายเป็นประจำ
- การปรับปรุงโครงสร้างโมเลกุล – การจำลองการจับกันของโมเลกุล (docking) สามารถใช้เพื่อทำนายตำแหน่งและทิศทางสัมพัทธ์ที่โมเลกุลจะจับกับโปรตีน (เรียกอีกอย่างว่าโหมดการจับหรือท่าทางการจับ) ข้อมูลนี้สามารถนำไปใช้ในการออกแบบโมเลกุลที่มีฤทธิ์แรงและมีความจำเพาะเจาะจงมากขึ้นได้
- การบำบัดทางชีวภาพ – การจับคู่ลิแกนด์โปรตีนยังสามารถใช้ในการทำนายสารมลพิษที่สามารถย่อยสลายได้ด้วยเอนไซม์[ 46 ] [ 47 ]
ดูเพิ่มเติม
- การออกแบบยา
- อัลกอริทึม Katchalski-Katzir
- รายชื่อระบบกราฟิกโมเลกุล
- การเชื่อมต่อโมเลกุลขนาดใหญ่
- กลศาสตร์โมเลกุล
- โครงสร้างโปรตีน
- การออกแบบโปรตีน
- ซอฟต์แวร์สำหรับการสร้างแบบจำลองกลศาสตร์โมเลกุล
- รายชื่อซอฟต์แวร์สำหรับการจับคู่โปรตีนกับลิแกนด์
- ซอฟต์แวร์ออกแบบโมเลกุล
- Docking@Home
- Exscalate4Cov
- อิเบอร์ซิวิส
- ฐานข้อมูล ZINC
- ผู้ค้นหาลูกค้าเป้าหมาย
- การฉายภาพยนตร์เสมือนจริง
- ฟังก์ชันการให้คะแนนสำหรับการเชื่อมต่อ
- การเชื่อมต่อขนาดใหญ่พิเศษ
ลิงก์ภายนอก
- Bikadi Z, Kovacs S, Demko L, Hazai E. "Molecular Docking Server - Ligand Protein Docking & Molecular Modeling" . Virtua Drug Ltd . สืบค้นเมื่อ2008-07-15 .
บริการทางอินเทอร์เน็ตที่คำนวณตำแหน่ง รูปทรงเรขาคณิต และพลังงานของโมเลกุลขนาดเล็กที่ทำปฏิกิริยากับโปรตีน
- Malinauskas T. "ขั้นตอนการติดตั้ง MGLTools 1.5.2 (AutoDockTools, Python Molecular Viewer และ Visual Programming Environment) บน Ubuntu Linux 8.04" . เก็บถาวรจากต้นฉบับเมื่อ 2009-02-26 . เรียกดูเมื่อ2008-07-15 .
- Docking@GRID ถูกเก็บถาวรเมื่อวันที่ 31 ธันวาคม 2019 ที่Wayback Machineโครงการการสุ่มตัวอย่างโครงสร้างและการเชื่อมต่อบนกริด : เป้าหมายหนึ่งคือการใช้งานอัลกอริทึมการเชื่อมต่อแบบกระจายภายในบนกริดการคำนวณ ดาวน์โหลดDocking@GRID เวอร์ชันโอเพนซอร์สสำหรับ Linux
- Click2Drug.org - สารบบเครื่องมือออกแบบยาโดยใช้คอมพิวเตอร์
- การเชื่อมต่อ ลิแกนด์กับตัวรับ (Ligand:Receptor Docking) ถูกเก็บถาวรเมื่อวันที่ 2 กุมภาพันธ์ 2019 ที่Wayback Machineโดยใช้ MOE (Molecular Operating Environment)