ชีวเคมีของโรคอัลไซเมอร์
ชีวเคมีของโรคอัลไซเมอร์ซึ่งเป็นสาเหตุที่พบบ่อยที่สุดของภาวะสมองเสื่อมยังไม่เป็นที่เข้าใจอย่างถ่องแท้โรคอัลไซเมอร์ (AD) ได้รับการระบุว่าเป็น โรค โปรตีโอพาธี : โรคที่เกิด จาก การพับตัวผิดปกติของโปรตีน อันเนื่องมาจากการสะสมของ โปรตีน อะไมลอยด์เบต้า (Aβ) ที่พับตัวผิดปกติในสมอง[ 1 ]อะไมลอยด์เบต้าเป็นเปปไทด์สั้นๆซึ่งเป็นผลพลอยได้ จากการ ย่อยสลายโปรตีน ที่ผิดปกติ ของโปรตีนทรานส์เมมเบรน อะไมลอยด์เบต้าพรีเคอร์เซอร์โปรตีน(APP) ซึ่งหน้าที่ของมันยังไม่ชัดเจน แต่คิดว่าเกี่ยวข้องกับการพัฒนาของเซลล์ประสาท[ 2 ] พรีเซนิลินเป็นส่วนประกอบของคอมเพล็กซ์โปรตีโอไลติกที่เกี่ยวข้องกับการประมวลผลและการย่อยสลาย APP [ 3 ] [ 4 ]
โมโนเมอร์ของอะไมลอยด์เบตาละลายได้และมีบริเวณสั้นๆ ของโครงสร้างทุติยภูมิแบบเบตาชีทและโพลีโพรลีน II เฮลิกซ์ ในสารละลาย[ 5 ]แม้ว่าส่วนใหญ่จะเป็นอัลฟาเฮลิก ซ์ ในเยื่อหุ้มเซลล์[ 6 ]อย่างไรก็ตาม ที่ความเข้มข้นสูงเพียงพอ พวกมันจะเกิดการเปลี่ยนแปลงโครงสร้าง อย่างมาก เพื่อสร้างโครงสร้างตติยภูมิ ที่อุดมไป ด้วยเบตาชีทซึ่งรวมตัวกันเพื่อสร้างเส้นใยอะไมลอยด์ [ 7 ] เส้นใยและ รูปแบบ โอลิโกเมอร์ของ Aβ เหล่านี้จะสะสมอยู่นอกเซลล์ประสาทในรูปแบบที่เรียกว่าคราบพลัคในผู้สูงอายุมีคราบพลัคหลายประเภท ได้แก่ คราบพลัคแบบกระจาย คราบพลั ค แบบ กะทัดรัด คราบพลัคแบบ มีแกนหรือ คราบพลัคแบบ นิวริติกรวมถึงการสะสมของ Aβ ในผนังของหลอดเลือดขนาดเล็กในสมองที่เรียกว่าโรคหลอดเลือดสมองอะไมลอยด์[ 8 ] [ 9 ]
AD ยังถือเป็น โรคที่เกี่ยวข้อง กับเทา (tauopathy)เนื่องจากการสะสมตัวที่ผิดปกติของโปรตีนเทาซึ่งเป็นโปรตีนที่เกี่ยวข้องกับไมโครทิวบูลที่แสดงออกในเซลล์ประสาทและทำหน้าที่ทำให้ไมโครทิวบูล ใน โครงสร้างเซลล์ มีเสถียรภาพ เช่นเดียวกับ โปรตีนที่เกี่ยวข้องกับไมโครทิวบูลส่วนใหญ่เทาจะถูกควบคุมโดยการฟอสโฟรีเลชัน ตามปกติ อย่างไรก็ตาม ในโรคอัลไซเมอร์ เทาที่มีการฟอสโฟรีเลชันมากเกินไปจะสะสมตัวเป็นเส้นใยเกลียวคู่[ 10 ]ซึ่งจะรวมตัวกันเป็นก้อนภายในเซลล์ประสาทที่เรียกว่าปมประสาท(neurofibrillary tangles)และนิวไรต์ ที่ผิดปกติ ซึ่งเกี่ยวข้องกับคราบอะไมลอยด์ แม้ว่าจะยังไม่ค่อยมีใครรู้เกี่ยวกับกระบวนการประกอบเส้นใย แต่การลดลงของ โปรตีน โพรลิลไอโซเมอเรสใน ตระกูล พาร์วูลินแสดงให้เห็นว่าเร่งการสะสมของเทาที่ผิดปกติ[ 11 ] [ 12 ]
การอักเสบของระบบประสาทมีส่วนเกี่ยวข้องในกระบวนการที่ซับซ้อนซึ่งนำไปสู่พยาธิสภาพและอาการของโรคอัลไซเมอร์ หลักฐานทางพยาธิวิทยาและทางคลินิกจำนวนมากบันทึกการเปลี่ยนแปลงทางภูมิคุ้มกันที่เกี่ยวข้องกับโรคอัลไซเมอร์ รวมถึงความเข้มข้นของไซโตไคน์ที่ก่อให้เกิดการอักเสบที่เพิ่มขึ้นในเลือดและน้ำไขสันหลัง [ 13 ] [ 14 ] ยังไม่เป็นที่เข้าใจอย่างถ่องแท้ว่าการเปลี่ยนแปลงเหล่านี้เป็นสาเหตุหรือผลที่ตามมาของโรคอัลไซเมอร์ แต่การอักเสบภายในสมอง รวมถึงปฏิกิริยาที่เพิ่มขึ้นของไมโครเกลีย ที่อยู่ประจำ ต่อการสะสมของอะไมลอยด์ มีส่วนเกี่ยวข้องกับการเกิดโรคและการดำเนินไปของโรคอัลไซเมอร์[ 15 ]ชีวเคมีส่วนใหญ่ที่ทราบเกี่ยวกับโรคอัลไซเมอร์ได้รับการถอดรหัสผ่านการวิจัยโดยใช้แบบจำลองการทดลองของโรคอัลไซเมอร์
พยาธิวิทยาประสาท
ใน ระดับ มหภาค AD มีลักษณะเฉพาะคือการสูญเสียเซลล์ประสาทและไซแนปส์ในเปลือกสมองและบริเวณใต้เปลือกสมองบางส่วน ส่งผลให้เกิดการฝ่อ อย่างรุนแรง ของบริเวณที่ได้รับผลกระทบ รวมถึงการเสื่อมสภาพในกลีบขมับและกลีบข้างและบางส่วนของเปลือกสมองส่วนหน้าและ ไจรัส ซิงกูเลต[ 16 ]
ทั้งคราบอะไมลอยด์และเส้นใยประสาทที่พันกันสามารถมองเห็นได้อย่างชัดเจนด้วยกล้องจุลทรรศน์ในสมองของผู้ป่วย AD [ 17 ]คราบอะไมลอยด์เป็นตะกอนหนาแน่นของโปรตีนและ วัสดุ เซลล์ที่ไม่ละลายน้ำ ส่วน ใหญ่อยู่นอกและรอบๆ เซลล์ประสาท ส่วนเส้นใยที่พันกันเป็นเส้นใยที่ไม่ละลายน้ำบิดเป็นเกลียวซึ่งสะสมอยู่ภายในเซลล์ประสาท แม้ว่าผู้สูงอายุหลายคนจะมีคราบอะไมลอยด์และเส้นใยที่พันกันอยู่บ้าง แต่สมองของผู้ป่วย AD จะมีคราบเหล่านี้ในปริมาณที่มากกว่ามากและอยู่ในตำแหน่งสมองที่แตกต่างกัน[ 18 ]
ลักษณะทางชีวเคมี
พื้นฐานในการทำความเข้าใจโรคอัลไซเมอร์คือเหตุการณ์ทางชีวเคมีที่นำไปสู่การสะสมของคราบอะไมลอยด์เบต้าและเส้นใยโปรตีนเทา ความสมดุลที่ละเอียดอ่อนของเอนไซม์ซี เคร เทสควบคุมการสะสมของอะไมลอยด์เบต้า เมื่อเร็วๆ นี้ มีการเน้นย้ำถึงความเชื่อมโยงระหว่างกิจกรรมของเซลล์ประสาทโคลินเนอร์จิกและกิจกรรมของอัลฟาซีเครเทส[ 19 ]ซึ่งสามารถยับยั้งการสะสมของโปรตีนอะไมลอยด์เบต้าในสมองของผู้ป่วยโรคอัลไซเมอร์ได้ โรคอัลไซเมอร์ได้รับการระบุว่าเป็น โรคที่เกิด จากการพับตัวผิดปกติของโปรตีนหรือโปรตีโอพาธีเนื่องจากมีการสะสมของโปรตีนอะไมลอยด์เบต้าที่พับตัวผิดปกติในสมองของผู้ป่วย AD [ 1 ]การสะสมของอะไมลอยด์เบต้าที่ผิดปกติสามารถตรวจพบได้ครั้งแรกโดยใช้การวิเคราะห์น้ำไขสันหลัง และต่อมาโดยใช้การตรวจเอกซเรย์คอมพิวเตอร์แบบโพซิตรอน (PET) [ 20 ]
แม้ว่า AD จะมีกลไกทางพยาธิสรีรวิทยาที่คล้ายคลึงกับโรคพรีออนแต่ก็ไม่สามารถแพร่กระจายในธรรมชาติได้เหมือนกับโรคพรีออน[ 21 ]การแพร่กระจายใดๆ ที่อาจเกิดขึ้นนั้นจำกัดอยู่เฉพาะเหตุการณ์ที่เกิดจากแพทย์ซึ่ง หายากมาก จากการบำบัดที่ได้รับจากผู้บริจาคซึ่งปัจจุบันเลิกใช้แล้ว[ 22 ]อะไมลอยด์เบต้า หรือเขียนว่า Aβ เป็นเปปไทด์ สั้นๆ ที่เป็น ผลพลอยได้จาก การย่อยสลาย โปรตีน ของ โปรตีนทราน ส์เมมเบรนอะไมลอยด์พรีเคอร์เซอร์ โปรตีน (APP) ซึ่งหน้าที่ของมันยังไม่ชัดเจน แต่เชื่อว่าเกี่ยวข้องกับการพัฒนาของเซลล์ประสาทพรีเซนิลินเป็นส่วนประกอบของคอมเพล็กซ์การย่อยสลายโปรตีนที่เกี่ยวข้องกับการประมวลผลและการย่อยสลาย APP [ 4 ] แม้ว่าโมโนเมอร์ ของอะไมลอยด์เบตา จะไม่เป็นอันตราย แต่พวกมันจะเกิดการเปลี่ยนแปลงโครงสร้าง อย่างมาก เมื่อมีความเข้มข้นสูงพอที่จะสร้างโครงสร้างตติยภูมิที่อุดม ไปด้วย เบตาชีทซึ่งจะรวมตัวกันเพื่อสร้างเส้นใยอะไมลอยด์[ 7 ]ที่สะสมอยู่นอกเซลล์ประสาทในรูปแบบหนาแน่นที่เรียกว่าคราบพลัคในผู้สูงอายุหรือคราบพลัคประสาทในรูปแบบการรวมตัวที่หนาแน่นน้อยกว่าที่เรียกว่าคราบพลัคแบบกระจายและบางครั้งก็อยู่ในผนังของหลอดเลือดขนาดเล็กในสมองในกระบวนการที่เรียกว่าโรคหลอดเลือดสมองจากอะไมลอยด์หรือโรคหลอดเลือดสมองจากคองโกฟิลิก
AD ยังถือเป็น โรคที่ เกี่ยวข้องกับโปรตีนเทาเนื่องจากการสะสมตัวที่ผิดปกติของโปรตีนเทา ซึ่งเป็นโปรตีนที่เกี่ยวข้องกับไมโครทิวบูลที่แสดงออกในเซลล์ประสาทและทำหน้าที่ทำให้ไมโครทิวบูล ใน โครงสร้างเซลล์ มี เสถียรภาพ เช่นเดียวกับโปรตีนที่เกี่ยวข้องกับไมโครทิวบูลส่วนใหญ่ โปรตีนเทาจะถูกควบคุมโดยการฟอสโฟรีเลชัน ตามปกติ อย่างไรก็ตาม ในผู้ป่วย AD โปรตีนเทาที่มีการฟอสโฟรีเลชันมากเกินไปจะสะสมตัวเป็นเส้นใยเกลียวคู่[ 10 ]ซึ่งจะรวมตัวกันเป็นก้อนภายในเซลล์ประสาทที่เรียกว่าปมประสาทเส้นใยและเส้นประสาท ที่เสื่อมสภาพ ซึ่งเกี่ยวข้องกับคราบอะไมลอยด์
ระดับของสารสื่อประสาทอะเซทิลโคลีน (ACh) ลดลง ระดับของสารสื่อประสาทอื่นๆ เช่น เซโรโทนินน อร์ เอพิเนฟรินและโซมาโตสแตตินก็มักจะลดลงเช่นกัน การเติม ACh ด้วยสารต้านโคลีนเอสเตอเรสเป็นวิธีการรักษาที่ได้รับการอนุมัติจาก FDA วิธีการทางเลือกในการกระตุ้นตัวรับ ACh ชนิด M1-M3 ด้วยสารกระตุ้นสังเคราะห์ที่มีอัตราการแยกตัวออกจากตัวรับที่ช้ากว่า ได้รับการเสนอให้เป็นโคลีนมิเมติกเจเนอเรชั่นถัดไปในโรคอัลไซเมอร์[ 23 ]
กลไกการเกิดโรค
แม้ว่าลักษณะทางเนื้อเยื่อวิทยาโดยรวมของ AD ในสมองจะได้รับการอธิบายอย่างดีแล้ว แต่ก็มีสมมติฐานที่แตกต่างกันหลายประการเกี่ยวกับสาเหตุหลัก สมมติฐานที่เก่าแก่ที่สุดประการหนึ่งคือสมมติฐานโคลินเนอร์จิกซึ่งชี้ให้เห็นว่าการขาดการส่งสัญญาณโคลินเนอร์จิกเป็นตัวเริ่มต้นความก้าวหน้าของโรค[ 24 ]ทฤษฎีปัจจุบันระบุว่าทั้งโปรตีนเทาที่พับผิดรูปภายในเซลล์และการรวมตัวของอะไมลอยด์เบต้าภายนอกเซลล์เป็นตัวเริ่มต้นกระบวนการที่นำไปสู่พยาธิสภาพของ AD [ 25 ] [ 26 ] สมมติฐานใหม่ที่มีศักยภาพเสนอปัจจัยทางเมตาบอลิ ซึม [ 27 ]ความผิดปกติของหลอดเลือด[ 28 ]การบุกรุกของไขมัน[ 29 ]และการอักเสบเรื้อรังในสมอง[ 30 ]เป็นปัจจัยที่ก่อให้เกิด AD สมมติฐานอะไมลอยด์เบต้าเกี่ยวกับการเริ่มต้นระดับโมเลกุลได้กลายเป็นที่แพร่หลายในหมู่นักวิจัยหลายคนจนถึงปัจจุบัน[ 31 ]สมมติฐานอะไมลอยด์และเทาเป็นที่ยอมรับกันอย่างกว้างขวางที่สุด
สมมติฐานเทา
สมมติฐานที่ว่าเทาเป็นปัจจัยหลักที่ก่อให้เกิดโรคนั้นมีพื้นฐานมาจากการสังเกตว่าการสะสมของคราบอะไมลอยด์ไม่ได้มีความสัมพันธ์ที่ดีกับการสูญเสียเซลล์ประสาท[ 32 ]กลไกของความเป็นพิษต่อระบบประสาทได้รับการเสนอขึ้นโดยอิงจากการสูญเสียโปรตีนเทาที่ทำให้ไมโครทิวบูลคงตัว ซึ่งนำไปสู่การเสื่อมสภาพของโครงสร้างเซลล์[ 33 ]อย่างไรก็ตาม ยังไม่มีข้อสรุปว่าการไฮเปอร์ฟอสโฟรีเลชันของเทาเกิดขึ้นก่อนหรือเป็นสาเหตุของการก่อตัวของกลุ่มเส้นใยเกลียวที่ผิดปกติ[ 34 ]การสนับสนุนสมมติฐานเทายังมาจากการมีอยู่ของโรคอื่นๆ ที่เรียกว่าเทาโอพาธีซึ่งโปรตีนชนิดเดียวกันนี้มีการพับตัวผิดปกติอย่างเห็นได้ชัด[ 35 ]อย่างไรก็ตาม นักวิจัยส่วนใหญ่สนับสนุนสมมติฐานทางเลือกที่ว่าอะไมลอยด์เป็นตัวการหลักที่ก่อให้เกิดโรค[ 34 ]
สมมติฐานอะไมลอยด์
สมมติฐานอะไมลอยด์ถูกเสนอขึ้นเนื่องจากยีนสำหรับสารตั้งต้นอะไมลอยด์เบต้า APP ตั้งอยู่บนโครโมโซม 21และผู้ป่วยที่มีภาวะไตรโซมี 21หรือที่รู้จักกันดีในชื่อ ดาวน์ ซินโดรมซึ่งมีสำเนายีนเพิ่มขึ้นหนึ่งชุดจะแสดงอาการคล้ายโรคอัลไซเมอร์เมื่ออายุ 40 ปี[ 36 ] [ 37 ]สมมติฐานอะไมลอยด์ชี้ให้เห็นถึงความเป็นพิษของเส้นใยอะไมลอยด์ที่รวมตัวกันอย่างสมบูรณ์ ซึ่งเชื่อว่าเป็นรูปแบบที่เป็นพิษของโปรตีนที่รับผิดชอบในการรบกวนสมดุล ของไอออนแคลเซียมในเซลล์ และทำให้เกิดอะพอพโทซิส [ 38 ] สมมติฐานนี้ได้รับการสนับสนุนจากการสังเกตว่าระดับที่สูงขึ้นของโปรตีนอะไมลอยด์เบต้าชนิดหนึ่งที่ทราบกันว่าก่อตัวเป็นเส้นใยได้เร็วกว่าในหลอดทดลองมีความสัมพันธ์กับการเริ่มมีอาการเร็วขึ้นและความบกพร่องทางสติปัญญาที่มากขึ้นในแบบจำลองหนู[ 39 ]และกับการวินิจฉัยโรคอัลไซเมอร์ในมนุษย์[ 40 ]อย่างไรก็ตาม กลไกสำหรับการไหลเข้าของแคลเซียมที่ถูกเหนี่ยวนำ หรือข้อเสนอสำหรับกลไกที่เป็นพิษต่อเซลล์ทางเลือกโดยเส้นใยที่เจริญเต็มที่นั้นยังไม่ชัดเจน
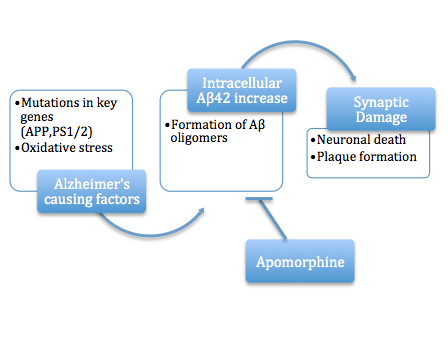
สมมติฐานอะไมลอยด์รูปแบบใหม่ล่าสุดระบุว่าชนิดที่เป็นพิษต่อเซลล์คือรูปแบบที่ผิดรูปของอะไมลอยด์เบต้าในระดับกลาง ซึ่งไม่ใช่ทั้งโมโนเมอร์ที่ละลายได้หรือพอลิเมอร์ที่รวมตัวกันอย่างสมบูรณ์ แต่ เป็นชนิด โอลิโกเมอร์อาจมีรูปร่างเป็นวงแหวนหรือรูปดาวที่มีช่องตรงกลาง[ 41 ]ซึ่งอาจกระตุ้นให้เกิดอะพอพโทซิสโดยการเจาะ เยื่อ หุ้มเซลล์[ 42 ]สมมติฐานช่องไอออนนี้ตั้งสมมติฐานว่าโอลิโกเมอร์ของ Aβ ที่ละลายได้และไม่เป็นเส้นใยจะก่อตัวเป็นช่องไอออนของเยื่อหุ้มเซลล์ ทำให้แคลเซียมไหลเข้าสู่เซลล์ประสาทอย่างไม่ควบคุม[ 43 ]ทางเลือกอื่นที่เกี่ยวข้องชี้ให้เห็นว่าโอลิโกเมอร์ทรงกลมที่อยู่เฉพาะที่กระบวนการเดนไดรต์และแอกซอนในเซลล์ประสาทเป็นชนิดที่เป็นพิษต่อเซลล์[ 44 ] [ 45 ]พบว่าสารรวมตัวก่อนเป็นเส้นใยสามารถทำลายเยื่อหุ้มเซลล์ได้[ 46 ]
สมมติฐานไซโตท็อกซิก-ไฟบริลนำเสนอเป้าหมายที่ชัดเจนสำหรับการพัฒนายา : ยับยั้งกระบวนการสร้างไฟบริล การพัฒนายา ในระยะเริ่มต้นส่วนใหญ่ มุ่งเน้นไปที่การยับยั้งนี้[ 47 ] [ 48 ] [ 49 ]ส่วนใหญ่ยังรายงานว่าช่วยลดความเป็นพิษต่อระบบประสาท แต่ทฤษฎีโอลิโกเมอร์ที่เป็นพิษจะบ่งชี้ว่าการป้องกันการประกอบโอลิโกเมอร์เป็นกระบวนการที่สำคัญกว่า[ 50 ] [ 51 ]หรือเป้าหมายที่ดีกว่านั้นอยู่ที่ต้นน้ำ เช่น การยับยั้งการประมวลผล APP ไปเป็นอะไมลอยด์เบต้า[ 52 ] ตัวอย่างเช่น พบว่าอะโพมอร์ฟีน ช่วยปรับปรุงการทำงานของความจำได้อย่างมีนัยสำคัญผ่านการทำภารกิจ Morris Water Mazeให้สำเร็จมากขึ้น[ 50 ]
- ละลายได้ภายในเซลล์ (o)Aβ42
เอกสารสองฉบับแสดงให้เห็นว่าโอลิโกเมอริก (o)Aβ42 (Aβ ชนิดหนึ่ง) ในรูปแบบที่ละลายได้ภายในเซลล์ ยับยั้งการส่งสัญญาณประสาท อย่างเฉียบพลัน ซึ่งเป็นพยาธิสภาพที่บ่งบอกถึงโรค AD (ในระยะเริ่มต้น) โดยการกระตุ้นเคซีนไคเนส 2 [ 53 ] [ 54 ]
สมมติฐานการอักเสบ
หลักฐานที่สอดคล้องกันชี้ให้เห็นว่าการตอบสนองการอักเสบที่ยั่งยืนในสมองเป็นคุณลักษณะการปรับเปลี่ยนหลักของพยาธิสภาพของโรคอัลไซเมอร์ และอาจเป็นปัจจัยปรับเปลี่ยนที่สำคัญในการเกิดโรคอัลไซเมอร์[ 55 ] [ 56 ]สมองของผู้ป่วยโรคอัลไซเมอร์แสดงให้เห็นเครื่องหมายหลายอย่างของการส่งสัญญาณการอักเสบที่เพิ่มขึ้น[ 57 ] [ 58 ] [ 59 ]สมมติฐานการอักเสบเสนอว่าการอักเสบที่เพิ่มขึ้นเรื้อรังในสมองเป็นองค์ประกอบสำคัญของกระบวนการอะไมลอยด์[ 60 ]ในระยะเริ่มต้นของโรคอัลไซเมอร์ และทำให้ความรุนแรงของโรคเพิ่มมากขึ้นในระยะหลังของโรคอัลไซเมอร์ Aβ มีอยู่ในสมองที่แข็งแรงและทำหน้าที่ทางสรีรวิทยาที่สำคัญในการฟื้นตัวจากการบาดเจ็บของเซลล์ประสาท การป้องกันการติดเชื้อ และการซ่อมแซมอุปสรรคเลือด-สมอง[ 61 ]อย่างไรก็ตาม ยังไม่ทราบว่าการผลิต Aβ เริ่มเกินความสามารถในการกำจัดของสมองและเริ่มต้นความก้าวหน้าของโรคอัลไซเมอร์ได้อย่างไร คำอธิบายที่เป็นไปได้คือ Aβ ทำให้ไมโครเกลียซึ่งเป็นเซลล์ภูมิคุ้มกันประจำสมอง ถูกกระตุ้นและหลั่งโมเลกุลส่งสัญญาณที่ก่อให้เกิดการอักเสบ เรียกว่าไซโตไคน์ซึ่งจะดึงดูดไมโครเกลียอื่นๆ ในบริเวณนั้น[ 62 ]ในขณะที่การกระตุ้นไมโครเกลียอย่างเฉียบพลัน เช่น ในการตอบสนองต่อการบาดเจ็บ เป็นประโยชน์และช่วยให้ไมโครเกลียกำจัด Aβ และเศษเซลล์อื่นๆ ผ่านการกลืนกิน แต่ไมโครเกลียที่ถูกกระตุ้นเรื้อรังจะมีประสิทธิภาพในการกำจัด Aβ ลดลง[ 55 ]แม้ว่าความสามารถในการกำจัด Aβ จะลดลง แต่ไมโครเกลียที่ถูกกระตุ้นยังคงหลั่งไซโตไคน์ที่ก่อให้เกิดการอักเสบ เช่น อินเตอร์ลิวคิน 1β และ 6 (IL-6, IL-1β) และปัจจัยเนื้องอกเนโครซิสอัลฟา (TNF-α) รวมถึงอนุมูลอิสระออกซิเจนซึ่งรบกวนการทำงานของไซแนปส์ที่ปกติ[ 63 ]และในที่สุดก็ทำให้เซลล์ประสาทตาย[ 64 ]การสูญเสียการทำงานของไซแนปส์และการตายของเซลล์ประสาทในภายหลังเป็นสาเหตุของความบกพร่องทางสติปัญญาและการสูญเสียปริมาตรในบริเวณสมองที่สำคัญซึ่งเกี่ยวข้องกับโรคอัลไซเมอร์[ 65 ] IL-1B, IL-6 และ TNF-α ทำให้เกิดการผลิต Aβ โอลิโกเมอร์เพิ่มขึ้น รวมถึงการไฮเปอร์ฟอสโฟรีเลชันของเทา ซึ่งนำไปสู่การกระตุ้นไมโครเกลียอย่างต่อเนื่องและสร้างกลไกป้อนกลับไปข้างหน้าซึ่งการผลิต Aβ เพิ่มขึ้นและการกำจัด Aβ ลดลงในที่สุดทำให้เกิดการก่อตัวของคราบ Aβ [ 66 ] [ 67 ]
สมมติฐานโคลินเนอร์จิกทางประวัติศาสตร์
สมมติฐานโคลินเนอร์จิกของการพัฒนา AD ได้รับการเสนอครั้งแรกในปี 1976 โดย Peter Davies และ AJF Maloney [ 68 ]สมมติฐานนี้อ้างว่าโรคอัลไซเมอร์เริ่มต้นจากการขาดการผลิตอะเซทิลโคลีน ซึ่งเป็น สารสื่อประสาทที่สำคัญการวิจัยการรักษาในช่วงแรกส่วนใหญ่ขึ้นอยู่กับสมมติฐานนี้ รวมถึงการฟื้นฟู "นิวเคลียสโคลินเนอร์จิก" ความเป็นไปได้ของการบำบัดด้วยการทดแทนเซลล์ได้รับการตรวจสอบบนพื้นฐานของสมมติฐานนี้ ยาต้านโรคอัลไซเมอร์รุ่นแรกทั้งหมดมีพื้นฐานมาจากสมมติฐานนี้และทำงานเพื่อรักษาอะเซทิลโคลีนโดยการยับยั้งอะเซทิลโคลีนเอสเตอเรส (เอนไซม์ที่สลายอะเซทิลโคลีน) ยาเหล่านี้แม้บางครั้งจะมีประโยชน์ แต่ก็ไม่ได้นำไปสู่การรักษาให้หายขาด ในทุกกรณี ยาเหล่านี้ทำหน้าที่เพียงรักษาอาการของโรคเท่านั้น และไม่ได้หยุดยั้งหรือย้อนกลับโรค ผลการศึกษาเหล่านี้และงานวิจัยอื่นๆ นำไปสู่ข้อสรุปว่า การขาดอะเซทิลโคลีนอาจไม่ได้เป็นสาเหตุโดยตรง แต่เป็นผลมาจากการทำลายเนื้อเยื่อสมองในวงกว้าง ซึ่งความเสียหายนั้นรุนแรงมากจนการบำบัดด้วยการทดแทนเซลล์อาจไม่สามารถทำได้จริง
ผลการค้นพบล่าสุดมุ่งเน้นไปที่ผลกระทบของโปรตีนที่พับผิดรูปและรวมตัวกัน ได้แก่ อะไมลอยด์เบต้าและเทา: ความผิดปกติ ของโปรตีนเทาอาจเป็นตัวเริ่มต้นของกระบวนการเกิดโรค จากนั้น การสะสมของ เบต้าอะไมลอยด์จะทำให้โรคดำเนินไป[ 34 ]
การบริโภคกลูโคส
สมองของมนุษย์เป็นหนึ่งในอวัยวะที่มีการเผาผลาญสูงที่สุดในร่างกาย และเผาผลาญกลูโคสจำนวนมากเพื่อผลิตพลังงานในเซลล์ในรูปของอะดีโนซีนไตรฟอสเฟต (ATP) [ 69 ]แม้ว่าจะมีความต้องการพลังงานสูง แต่สมองก็มีความยืดหยุ่นค่อนข้างจำกัดในการใช้สารตั้งต้นเพื่อผลิตพลังงาน และพึ่งพากลูโคสที่ไหลเวียนอยู่เกือบทั้งหมดเพื่อตอบสนองความต้องการพลังงาน[ 70 ]การพึ่งพากลูโคสนี้ทำให้สมองมีความเสี่ยงหากการจัดหากลูโคสถูกขัดจังหวะ หรือหากความสามารถในการเผาผลาญกลูโคสของสมองบกพร่อง หากสมองไม่สามารถผลิต ATP ได้ ซินแนปส์จะไม่สามารถคงอยู่ได้ และเซลล์จะไม่สามารถทำงานได้ ซึ่งในที่สุดจะนำไปสู่ความบกพร่องทางสติปัญญา[ 70 ]
การศึกษาภาพแสดงให้เห็นว่าการใช้กลูโคสในสมองของผู้ป่วยโรคอัลไซเมอร์ลดลงในช่วงเริ่มต้นของโรค ก่อนที่จะมีสัญญาณทางคลินิกของความบกพร่องทางสติปัญญา การลดลงของการเผาผลาญกลูโคส นี้ จะแย่ลงเมื่ออาการทางคลินิกพัฒนาขึ้นและโรคดำเนินไป[ 71 ] [ 72 ] การศึกษาพบว่าการเผาผลาญกลูโคสในสมองลดลง 17%-24% ในผู้ป่วยโรคอัลไซเมอร์ เมื่อเทียบกับกลุ่มควบคุมที่มีอายุใกล้เคียงกัน[ 73 ]การศึกษาภาพจำนวนมากได้ยืนยันการสังเกตนี้ตั้งแต่นั้นมา
อัตราการเผาผลาญกลูโคสในสมองที่ต่ำกว่าปกติพบได้ในรูปแบบลักษณะเฉพาะในสมองของผู้ป่วยโรคอัลไซเมอร์ โดยเฉพาะอย่างยิ่งในบริเวณซีงกูเลตด้านหลัง คอร์เทกซ์ข้างขมับ คอร์เทกซ์ขมับ และคอร์เทกซ์หน้าผาก เชื่อกันว่าบริเวณสมองเหล่านี้ควบคุมความจำและการรับรู้ หลายด้าน รูปแบบการเผาผลาญนี้สามารถทำซ้ำได้ และยังได้รับการเสนอให้เป็นเครื่องมือวินิจฉัยโรคอัลไซเมอร์อีกด้วย ยิ่งไปกว่านั้น การเผาผลาญกลูโคสในสมองที่ลดลง (DCGM) ยังมีความสัมพันธ์กับความหนาแน่นของคราบพลัคและความบกพร่องทางสติปัญญาในผู้ป่วยที่มีโรคขั้นรุนแรงมากขึ้น[ 73 ] [ 74 ]
การเผาผลาญกลูโคสในสมองที่ลดลง (DCGM) อาจไม่ใช่เพียงผลจากการสูญเสียเซลล์สมองเท่านั้น เนื่องจากเกิดขึ้นในผู้ป่วยที่ไม่มีอาการแต่มีความเสี่ยงต่อโรคอัลไซเมอร์ เช่น ผู้ป่วยที่เป็นโฮโมไซกัสสำหรับ ยีน อะโพลิโปโปรตีนอี สายพันธุ์เอปไซลอน 4 (APOE4 ซึ่งเป็นปัจจัยเสี่ยงทางพันธุกรรมของโรคอัลไซเมอร์) รวมถึงโรคอัลไซเมอร์ที่ถ่ายทอดทางพันธุกรรมด้วย[ 75 ]เนื่องจาก DCGM เกิดขึ้นก่อนการเปลี่ยนแปลงทางคลินิกและพยาธิวิทยาอื่นๆ จึงไม่น่าจะเกิดจากการสูญเสียเซลล์จำนวนมากที่พบในโรคอัลไซเมอร์[ 70 ]
ในการศึกษาภาพถ่ายที่เกี่ยวข้องกับผู้ใหญ่ตอนต้นที่เป็นพาหะ APOE4 ซึ่งไม่มีสัญญาณของความบกพร่องทางสติปัญญา พบว่าการเผาผลาญกลูโคสในสมองลดลง (DCGM) ในบริเวณสมองเดียวกันกับผู้สูงอายุที่เป็นโรคอัลไซเมอร์[ 75 ]อย่างไรก็ตาม DCGM ไม่ได้พบเฉพาะในพาหะ APOE4 เท่านั้น เมื่อได้รับการวินิจฉัยว่าเป็นโรคอัลไซเมอร์แล้ว DCGM จะเกิดขึ้นในจีโนไทป์ APOE3/E4, APOE3/E3 และ APOE4/E4 [ 76 ]ดังนั้น DCGM จึงเป็นตัวบ่งชี้ทางชีวภาพของ การเผาผลาญ สำหรับสภาวะของโรค[ 77 ]
การส่งสัญญาณอินซูลิน
ในช่วงทศวรรษที่ผ่านมา มีการค้นพบความเชื่อมโยงระหว่างโรคอัลไซเมอร์และโรคเบาหวาน เนื่องจากพบว่าภาวะดื้อต่ออินซูลินซึ่งเป็นลักษณะเด่นของโรคเบาหวานก็พบได้ในสมองของผู้ป่วยโรคอัลไซเมอร์ เช่นกัน [ 78 ] สารประกอบ อะไมลอยด์-เบตา แบบโอลิโกเมอริกที่เป็นพิษต่อระบบ ประสาทจะลดการแสดงออกของตัวรับอินซูลินบนพื้นผิวเซลล์ประสาท[ 79 ]และทำให้การส่งสัญญาณอินซูลินของเซลล์ประสาทหยุดชะงัก[ 78 ]มีการเสนอแนะว่าแกงกลิโอไซด์ ของเซลล์ประสาท ซึ่งมีส่วนร่วมในการสร้างไมโครโดเมนของไขมัน ในเยื่อหุ้ม เซลล์ ช่วยอำนวยความสะดวกในการกำจัดตัวรับอินซูลินออกจากพื้นผิวเซลล์ประสาทที่เกิดจากอะไมลอยด์-เบตา[ 80 ]ในโรคอัลไซเมอร์ สารประกอบอะไมลอยด์-เบตาแบบโอลิโกเมอริกจะกระตุ้นการส่งสัญญาณTNF-α [ 78 ]การกระตุ้น c-Jun N-terminal kinase โดย TNF-α จะกระตุ้น kinase ที่เกี่ยวข้องกับความเครียดและส่งผลให้เกิด การฟอสโฟรีเลชันของ ซีรีน ใน IRS-1 ซึ่งจะปิดกั้นการส่งสัญญาณอินซูลินในขั้นตอนถัดไป[ 78 ] [ 81 ] [ 82 ]ภาวะดื้อต่ออินซูลินที่เกิดขึ้นส่งผลให้เกิดความบกพร่องทางสติปัญญา ดังนั้น การเพิ่มความไวต่ออินซูลินและการส่งสัญญาณของเซลล์ประสาทอาจเป็นแนวทางการรักษาแบบใหม่ในการรักษาโรคอัลไซเมอร์[ 83 ] [ 84 ]
ความเครียดออกซิเดชัน
ความเครียดจากออกซิเดชันกำลังกลายเป็นปัจจัยสำคัญในการเกิดโรค AD [ 85 ] เชื่อกันว่าการผลิตสารออกซิเจนที่ว่องไว (ROS) มากเกินไปมีบทบาทสำคัญในการสะสมและการตกตะกอนของ อะไมลอยด์เบต้าใน AD [ 86 ] สมองของผู้ป่วย AD มีระดับความเสียหายของ DNA จากออกซิเดชัน สูงขึ้น ทั้งในDNA นิวเคลียร์และ ไมโทคอนเดรีย แต่ DNA ไมโทคอนเดรียมีระดับสูงกว่า DNA นิวเคลียร์ประมาณ 10 เท่า[ 87 ] ไมโทคอนเดรียที่เสื่อมสภาพตามอายุอาจเป็นปัจจัยสำคัญในการเกิดการเสื่อมของระบบประสาทใน AD [ 86 ] แม้แต่บุคคลที่มีความบกพร่องทางสติปัญญาเล็กน้อยซึ่งเป็นระยะระหว่างการแก่ตัวตามปกติและภาวะสมองเสื่อมระยะเริ่มต้น ก็มีความเสียหายจากออกซิเดชันเพิ่มขึ้นใน DNA นิวเคลียร์และไมโทคอนเดรีย ในสมอง [ 88 ] (ดูสมองที่เสื่อมสภาพตามอายุ ) การแตกของดีเอ็นเอแบบสองสาย (DSBs) ที่เกิด ขึ้นตามธรรมชาติในเซลล์มนุษย์ส่วนใหญ่เกิดจากการแตกของสายเดี่ยวที่เกิดจากกระบวนการต่างๆ รวมถึงกิจกรรมของสารออกซิเจนที่ว่องไว โทโปไอโซเมอเรส และการไฮโดรไลซิสเนื่องจากความผันผวนของอุณหภูมิ[ 89 ] ในเซลล์ประสาท DSBs เกิดขึ้นจากโทโปไอโซเมอเรสชนิดที่ 2 ซึ่งเป็นส่วนหนึ่งของกระบวนการทางสรีรวิทยาของการสร้างความทรงจำ[ 90 ] DSBs พบได้ทั้งในเซลล์ประสาทและแอสโทรไซต์ในฮิปโปแคมปัส ของมนุษย์ที่เสียชีวิตแล้ว ของผู้ป่วย AD ในระดับที่สูงกว่าในบุคคลที่ไม่เป็น AD [ 91 ] AD เกี่ยวข้องกับการสะสมของ DSBs ในเซลล์ประสาทและแอสโทรไซต์ในฮิปโปแคมปัสและคอร์เทกซ์ส่วนหน้าตั้งแต่ระยะเริ่มต้นเป็นต้นไป[ 92 ] DSBs เพิ่มขึ้นในบริเวณใกล้เคียงกับคราบอะไมลอยด์ในฮิปโปแคมปัส ซึ่งบ่งชี้ถึงบทบาทที่เป็นไปได้ของ Aβ ในการสะสม DSB หรือในทางกลับกัน[ 91 ] กลไกหลักในการซ่อมแซมการแตกของสายคู่ DNA คือการเชื่อมต่อปลายที่ไม่เหมือนกัน (NHEJ) ซึ่งเป็นกลไกที่ใช้คอมเพล็กซ์ DNA-dependent protein kinase (DNA-PK) กิจกรรมการเชื่อมต่อปลายและระดับโปรตีนของหน่วยย่อยเร่งปฏิกิริยาของ DNA-PK ต่ำกว่าอย่างมีนัยสำคัญในสมองของผู้ป่วย AD เมื่อเทียบกับสมองปกติ[ 93 ]
สมมติฐานเกี่ยวกับคอเลสเตอรอล
สมมติฐานคอเลสเตอรอลเป็นการรวมกันของสมมติฐานอะไมลอยด์ สมมติฐานเทา และอาจรวมถึงสมมติฐานการอักเสบด้วย พบว่าคอเลสเตอรอลอยู่เหนือการผลิตอะไมลอยด์และเทา[ 94 ]คอเลสเตอรอลถูกผลิตในแอสโทรไซต์และส่งไปยังเซลล์ประสาท ซึ่งจะกระตุ้นการผลิตอะไมลอยด์ผ่านกระบวนการที่เรียกว่าการนำเสนอซับสเตรตกระบวนการนี้ต้องใช้ apoE การควบคุมการผลิตเทาของคอเลสเตอรอลยังไม่เป็นที่เข้าใจดีนัก แต่การกำจัดเอนไซม์สังเคราะห์คอเลสเตอรอล SREBP2 ทำให้การฟอสโฟรีเลชันของเทาลดลง[ 94 ]ภูมิคุ้มกันโดยกำเนิดกระตุ้นการสังเคราะห์คอเลสเตอรอลและเซลล์จะดูดซับคอเลสเตอรอล[ 95 ]สันนิษฐานว่าเซลล์ในสมองจะตายเมื่ออายุมากขึ้นและสิ่งนี้จะกระตุ้นภูมิคุ้มกันโดยกำเนิด จำเป็นต้องมีการศึกษาเพิ่มเติมเพื่อเชื่อมโยงสมมติฐานการอักเสบกับการสังเคราะห์คอเลสเตอรอลในสมองโดยตรง
สมมติฐานการบุกรุกของไขมัน
แบบจำลองการบุกรุกของไขมัน (LIM) เป็นสมมติฐาน[ 96 ] [ 97 ]สำหรับโรคอัลไซเมอร์ที่ตีพิมพ์ในปี 2022 ซึ่งกล่าวว่าโรคอัลไซเมอร์เป็นผลมาจากการบุกรุกของไขมันจากภายนอกเข้าสู่สมอง ภายหลังความเสียหายต่ออุปสรรคเลือด-สมอง (BBB) LIM ให้คำอธิบายที่ครอบคลุมเกี่ยวกับพยาธิสภาพทางประสาทที่สังเกตได้ซึ่งเกี่ยวข้องกับโรคนี้ รวมถึงความผิดปกติของไขมันที่ Alois Alzheimer อธิบายไว้เป็นครั้งแรก และอธิบายถึงปัจจัยเสี่ยงที่หลากหลายที่ระบุได้ในปัจจุบันเกี่ยวกับโรคอัลไซเมอร์ (รวมถึงอายุมาก, ApoE4, Aβ, การบาดเจ็บที่สมอง, ความดันโลหิตสูง, การสูบบุหรี่, โรคเบาหวานชนิดที่ 2, โรคอ้วน, แอลกอฮอล์, ความเครียด และการนอนหลับไม่เพียงพอ) ซึ่งส่วนใหญ่เกี่ยวข้องกับความเสียหายต่อ BBB ด้วย[ 98 ] [ 99 ] [ 100 ] [ 101 ] [ 102 ] [ 103 ] [ 104 ] [ 105 ]
LIM สามารถมองได้ว่าเป็นการพัฒนาต่อยอดจากสมมติฐานคอเลสเตอรอล และรวมเอาและขยายสมมติฐานอะไมลอยด์ ซึ่งเป็นคำอธิบายที่โดดเด่นในปัจจุบันของโรคนี้ โดยย้อนกลับไปหนึ่งขั้นตอนเพื่อโต้แย้งว่าสาเหตุของคราบอะไมลอยด์ เส้นใยประสาท/เทาที่พันกัน และลักษณะอื่นๆ อีกมากมายของโรค คือการบุกรุกของไลโปโปรตีนความหนาแน่นต่ำ (LDL) และคอเลสเตอรอลชนิด "ไม่ดี" รูปแบบอื่นๆ พร้อมกับกรดไขมันอิสระ (FFAs) เข้าสู่สมอง หลังจากการแตกตัวของ BBB ไขมันดังกล่าวโดยปกติจะถูกกีดกันออกจากสมองโดย BBB [ 106 ] [ 107 ]
LIM โต้แย้งว่าการไหลเข้าของ 'คอเลสเตอรอลที่ไม่ดี' เป็นสาเหตุหลักของ Aβ ที่มากเกินไป การก่อตัวของคราบพลัค และเส้นใยประสาท/เทาที่พันกันในโรคอัลไซเมอร์ชนิดเริ่มช้า (LOAD) เนื่องจากการเปลี่ยนแปลงใน องค์ประกอบ ของแพไขมันและ การขนส่ง เอนโดโซม-ไลโซโซมซึ่งสอดคล้องกับหลักฐานจำนวนมากที่แสดงให้เห็นถึงความสัมพันธ์ของคอเลสเตอรอลที่มากเกินไปกับการผลิต Aβ ที่เพิ่มขึ้น คราบอะไมลอยด์[ 108 ] [ 109 ]และเส้นใยประสาท/เทาที่พันกัน[ 110 ] [ 111 ]
เชื่อกันว่าคราบโปรตีนและเส้นใยโปรตีนที่พันกันเป็นสาเหตุของการสูญเสียความจำในโรคอัลไซเมอร์ อย่างไรก็ตาม สมองของผู้ป่วยอัลไซเมอร์ไม่ได้แสดงคราบโปรตีนหรือเส้นใยโปรตีนที่พันกันเสมอไป และคราบโปรตีนและเส้นใยโปรตีนที่พันกันก็ไม่ได้นำไปสู่โรคอัลไซเมอร์เสมอไป ดังนั้น LIM จึงเสนอว่ากรดไขมันอิสระ (FFAs) มากกว่าโปรตีนอะไมลอยด์เบตา (Aβ) ที่เกิดจากคอเลสเตอรอล อาจเป็นตัวขับเคลื่อนหลักของโรคอัลไซเมอร์ กรดไขมันอิสระสามารถอธิบายลักษณะทั่วไปทั้งหมดของโรคอัลไซเมอร์ได้ รวมถึงภาวะความจำเสื่อม การหยุดชะงักของไซแนปส์ การอักเสบของระบบประสาท การหดตัวของสมอง การหยุดชะงักของนาฬิกาชีวภาพ การเปลี่ยนแปลงการผลิตพลังงานในสมองจากกลูโคสไปเป็นคีโตน ความเป็นพิษของไมโทคอนเดรีย และความเครียดออกซิเดชันภายในเซลล์ประสาท LIM โต้แย้งว่าผลกระทบของกรดไขมันอิสระอาจเป็นสาเหตุของการสูญเสียความจำส่วนใหญ่ในโรคอัลไซเมอร์ นอกเหนือจากความสับสนเชิงพื้นที่ การนอนหลับไม่ปกติ และบางครั้งอาการหวาดระแวงที่เกี่ยวข้องกับโรคนี้ด้วย
แบบจำลองการบุกรุกของไขมัน (Lipid Invasion Model หรือ LIM) เป็นแบบจำลองเดียวของโรคอัลไซเมอร์ที่สามารถอธิบายได้ทั้งคราบพลัคและเส้นใยประสาท/กลุ่มโปรตีนเทาที่พบได้ทั่วไปในโรคอัลไซเมอร์ชนิดเริ่มช้า (ซึ่งคิดเป็น 95% ของผู้ป่วยโรคอัลไซเมอร์ทั้งหมด) รวมถึงลักษณะมาตรฐานอื่นๆ ของโรคอัลไซเมอร์ด้วย นอกจากนี้ยังอธิบายได้ว่าทำไมโรคอัลไซเมอร์จึงส่งผลกระทบต่อผู้สูงอายุมากกว่ากลุ่มอื่นๆ และทำไมจึงพบได้บ่อยในนักกีฬาที่ต้องมีการปะทะกัน โดยการกล่าวว่าสาเหตุหลักของโรคอัลไซเมอร์คือความเสียหายต่ออุปสรรคเลือดสมอง (BBB) และการบุกรุกของไขมันที่เป็นอันตรายในภายหลัง แบบจำลองนี้จึงให้ข้อมูลเชิงลึกใหม่เกี่ยวกับสาเหตุพื้นฐานของโรคอัลไซเมอร์ และแนวทางใหม่ๆ ที่เป็นไปได้สำหรับการรักษาโรคนี้ LIM อาจให้ข้อมูลเชิงลึกเกี่ยวกับภาวะสมองเสื่อมและโรคทางระบบประสาทอื่นๆ เช่น โรคพาร์กินสัน และโรค ALS/โรคเซลล์ประสาทสั่งการเสื่อม (Motor Neurone Disease) ด้วย
สมมติฐานรีลิน
การศึกษาในปี 1994 [ 112 ]แสดงให้เห็นว่า การเปลี่ยนแปลง ของไอโซพรีนอยด์ในโรคอัลไซเมอร์นั้นแตกต่างจากที่เกิดขึ้นในระหว่างการแก่ตัวตามปกติ และโรคนี้จึงไม่สามารถถือได้ว่าเป็นผลมาจากการแก่ตัวก่อนวัยในระหว่างการแก่ตัวสมองของมนุษย์จะแสดงให้เห็นถึงการเพิ่มขึ้นอย่างต่อเนื่องของระดับโดลิคอลการลดลงของระดับยูบิควิโนนแต่ความเข้มข้นของคอเลสเตอรอล และโดลิชิลฟอสเฟต ยัง คงค่อนข้างคงที่ ในโรคอัลไซเมอร์ สถานการณ์กลับกัน โดยมีระดับโดลิคอล ลดลง และระดับยูบิควิโนน เพิ่มขึ้น ความเข้มข้นของโดลิชิลฟอสเฟตก็เพิ่มขึ้นเช่นกัน ในขณะที่ คอเลสเตอรอลยังคงคงที่ การเพิ่มขึ้นของโดลิชิลฟอสเฟตซึ่งเป็นตัวนำน้ำตาลอาจสะท้อนถึงอัตราการไกลโคซิเลชัน ที่เพิ่มขึ้น ในสมองที่เป็นโรค และการเพิ่มขึ้นของยูบิควิโนนซึ่งเป็นสารต้านอนุมูล อิสระภายในร่างกาย เป็นความพยายามที่จะปกป้องสมองจากความเครียดจากออกซิเดชันเช่น ที่เกิดจากการเกิดปฏิกิริยาออกซิเดชันของไขมัน[ 112 ] Ropren ซึ่งระบุไว้ก่อนหน้านี้ในรัสเซีย มีฤทธิ์ปกป้องระบบประสาทในแบบจำลองโรคอัลไซเมอร์ในหนู[ 113 ] [ 114 ]
สมมติฐานที่ค่อนข้างใหม่ซึ่งอิงจากการทดลองในสัตว์ฟันแทะเป็นหลัก เชื่อมโยงการเริ่มต้นของโรคอัลไซเมอร์กับการทำงานที่ลดลงของโปรตีนรีลิน ซึ่งเป็นโปรตีนนอกเซลล์ขนาดใหญ่ การลดลง ของรีลินในเยื่อหุ้มสมองส่วนเอนโทไรนัลของมนุษย์ซึ่งเป็นบริเวณที่โรคมักเริ่มต้นขึ้นนั้นเห็นได้ชัด[ 115 ]ในขณะเดียวกันก็มีรายงานการเพิ่มขึ้นของระดับรีลินเพื่อชดเชยในโครงสร้างสมองอื่นๆ ของผู้ป่วยด้วย[ 116 ]ที่สำคัญอย่างยิ่ง การแสดงออกของรีลินที่มากเกินไปช่วยฟื้นฟูความสามารถทางปัญญาของหนูทดลองที่เป็นแบบจำลองโรคอัลไซเมอร์[ 117 ]และหนูทดลองที่แสดงออกโปรตีน τ มากเกินไป[ 118 ]แบบจำลองระดับวงจรล่าสุดได้เสนอกลไกที่การลดลงของรีลินนำไปสู่การเสื่อมถอยของความทรงจำแบบเหตุการณ์ ในระยะเริ่มต้น ซึ่งเป็นการวางรากฐานทางทฤษฎีของสมมติฐานรีลิน[ 119 ]
สมมติฐานความไม่เสถียรของยีนขนาดใหญ่
การวิเคราะห์ทางชีวสารสนเทศในปี 2017 [ 120 ]เผยให้เห็นว่ายีนของมนุษย์ขนาดใหญ่มากมีการแสดงออกมากเกินไปอย่างมีนัยสำคัญในสมองและมีส่วนร่วมในโครงสร้างโพสต์ไซแนปส์ ยีนเหล่านี้ยังอุดมไปด้วยคำศัพท์ Gene Ontology (GO) เกี่ยวกับการยึดเกาะของเซลล์ และมักจะแมปไปยังตำแหน่งเปราะบางของโครโมโซม[ 121 ]ผลิตภัณฑ์ยีนเสี่ยงต่อโรคอัลไซเมอร์ที่รู้จักส่วนใหญ่ รวมถึงโปรตีนสารตั้งต้นอะไมลอยด์ (APP) และแกมมา-ซีเครเทส ตลอดจนตัวรับ APOE และตำแหน่งเสี่ยง GWAS มีส่วนร่วมในกลไกการยึดเกาะของเซลล์ที่คล้ายคลึงกัน สรุปได้ว่าการทำงานผิดปกติของการยึดเกาะของเซลล์และไซแนปส์เป็นหัวใจสำคัญของการเกิดโรคอัลไซเมอร์ และความไม่เสถียรของการกลายพันธุ์ของยีนการยึดเกาะไซแนปส์ขนาดใหญ่อาจเป็นตัวกระตุ้นทางสาเหตุของการหยุดชะงักของการส่งสัญญาณประสาทและการสูญเสียไซแนปส์ในสมองที่แก่ชรา ตัวอย่างเช่น สมมติฐานนี้อธิบายตำแหน่งเสี่ยงของยีน APOE ที่เกี่ยวข้องกับโรคอัลไซเมอร์ในบริบทของการส่งสัญญาณผ่านตัวรับไลโปโปรตีนขนาดใหญ่ LRP1b ซึ่งเป็นยีนยับยั้งเนื้องอกขนาดใหญ่ที่มีการแสดงออกเฉพาะในสมอง และยังอยู่ในตำแหน่งเปราะบางของโครโมโซมที่ไม่เสถียร สมมติฐานเกี่ยวกับความไม่เสถียรของยีนขนาดใหญ่ทำให้กลไกความเสียหายของดีเอ็นเอเป็นศูนย์กลางของพยาธิสรีรวิทยาของโรคอัลไซเมอร์