เทคโนโลยีทรานสคริปโตมิกส์
เทคโนโลยีทรานสคริปโตมิกส์เป็นเทคนิคที่ใช้ในการศึกษาทรานสค ริปโตมของสิ่งมีชีวิต ซึ่งเป็นผลรวมของ ทรานสคริปต์ RNAทั้งหมดของสิ่งมีชีวิตนั้น ข้อมูลของสิ่งมีชีวิตถูกบันทึกไว้ใน DNA ของจีโนมและแสดงออกผ่านกระบวนการถอดรหัสในที่นี้mRNAทำหน้าที่เป็นโมเลกุลตัวกลางชั่วคราวในเครือข่ายข้อมูล ในขณะที่RNA ที่ไม่เข้ารหัสทำหน้าที่อื่นๆ ที่หลากหลาย ทรานสคริปโตมจะบันทึกภาพรวมของทรานสคริปต์ทั้งหมดที่มีอยู่ในเซลล์ ณ เวลาใดเวลา หนึ่ง เทคโนโลยีทรานสคริปโตมิกส์ให้ข้อมูลโดยละเอียดเกี่ยวกับกระบวนการต่างๆ ในเซลล์ที่กำลังทำงานอยู่และกระบวนการที่อยู่ในสภาวะหยุดนิ่ง ความท้าทายที่สำคัญในชีววิทยาระดับโมเลกุลคือการทำความเข้าใจว่าจีโนมเดียวสามารถก่อให้เกิดเซลล์ที่หลากหลายได้อย่างไร อีกความท้าทายหนึ่งคือการควบคุมการแสดงออกของยีน
ความพยายามครั้งแรกในการศึกษาจีโนมทั้งหมดเริ่มต้นขึ้นในช่วงต้นทศวรรษ 1990 ความก้าวหน้าทางเทคโนโลยีที่เกิดขึ้นหลังจากนั้นตั้งแต่ปลายทศวรรษ 1990 ได้เปลี่ยนแปลงวงการนี้อย่างต่อเนื่องและทำให้จีโนมกลายเป็นสาขาวิชาที่แพร่หลายในวิทยาศาสตร์ชีวภาพมีเทคนิคสำคัญสองอย่างในปัจจุบัน ได้แก่ไมโครอาร์เรย์ซึ่งวัดปริมาณลำดับที่กำหนดไว้ล่วงหน้า และRNA-Seqซึ่งใช้การจัดลำดับแบบความเร็วสูงเพื่อบันทึกทรานสคริปต์ทั้งหมด เมื่อเทคโนโลยีดีขึ้น ปริมาณข้อมูลที่ได้จากการทดลองจีโนมแต่ละครั้งก็เพิ่มขึ้น ส่งผลให้วิธีการวิเคราะห์ข้อมูลได้รับการปรับปรุงอย่างต่อเนื่องเพื่อวิเคราะห์ข้อมูลปริมาณมากได้อย่างแม่นยำและมีประสิทธิภาพมากขึ้น ฐานข้อมูลจีโนมจึงมีขนาดใหญ่ขึ้นและมีประโยชน์มากขึ้นเรื่อยๆ เนื่องจากนักวิจัยยังคงรวบรวมและแบ่งปันจีโนมอย่างต่อเนื่อง การตีความข้อมูลที่มีอยู่ในจีโนมโดยปราศจากความรู้จากการทดลองก่อนหน้านี้แทบจะเป็นไปไม่ได้เลย
การวัดการแสดงออกของยีน ในสิ่งมีชีวิตใน เนื้อเยื่อหรือสภาวะต่างๆหรือในช่วงเวลาต่างๆ จะให้ข้อมูลเกี่ยวกับการควบคุม ยีน และเผยรายละเอียดทางชีววิทยาของสิ่งมีชีวิต นอกจากนี้ยังสามารถใช้เพื่ออนุมานหน้าที่ของ ยีน ที่ไม่เคยมีการระบุ มาก่อนได้ การวิเคราะห์ทรานสคริปโตมช่วยให้เข้าใจการเปลี่ยนแปลงการแสดงออกของยีนในสิ่งมีชีวิตต่างๆ ได้ดียิ่งขึ้น และมีบทบาทสำคัญในการทำความเข้าใจโรค ในมนุษย์ การวิเคราะห์การแสดงออกของยีนโดยรวมช่วยให้สามารถตรวจจับแนวโน้มที่ประสานกันในวงกว้าง ซึ่งไม่สามารถแยกแยะได้ด้วย การทดสอบแบบเจาะจงมากกว่า
ประวัติศาสตร์
ทรานสคริปโตมิกส์มีลักษณะเด่นคือการพัฒนาเทคนิคใหม่ๆ ที่ได้กำหนดนิยามใหม่ของสิ่งที่เป็นไปได้ทุกๆ ทศวรรษ และทำให้เทคโนโลยีเดิมล้าสมัยไป ความพยายามครั้งแรกในการเก็บรวบรวมทรานสคริปโตมของมนุษย์บางส่วนได้รับการตีพิมพ์ในปี 1991 และรายงานลำดับ mRNA จำนวน 609 ลำดับจากสมองของมนุษย์[ 2 ]ในปี 2008 มีการตีพิมพ์ทรานสคริปโตมของมนุษย์สองชุด ซึ่งประกอบด้วยลำดับที่ได้จากทรานสคริปต์หลายล้านลำดับ ครอบคลุมยีน 16,000 ยีน[ 3 ] [ 4 ]และภายในปี 2015 มีการตีพิมพ์ทรานสคริปโตมสำหรับบุคคลหลายร้อยคน[ 5 ] [ 6 ]ปัจจุบันมีการสร้าง ทรานสคริ ปโตมของสภาวะโรค ต่างๆ เนื้อเยื่อหรือแม้แต่เซลล์ เดี่ยวๆ เป็นประจำ [ 6 ] [ 7 ] [ 8 ]การเติบโตอย่างรวดเร็วของทรานสคริปโตมิกส์นี้เกิดจากการพัฒนาเทคโนโลยีใหม่ๆ อย่างรวดเร็วด้วยความไวและความประหยัดที่ดียิ่งขึ้น[ 9 ] [ 10 ] [ 11 ] [ 12 ]
ก่อนยุคทรานสคริปโตมิกส์
มีการศึกษา การถอดรหัสแต่ละ รายการ มาหลายทศวรรษก่อนที่จะมีวิธีการถอดรหัสทางพันธุกรรมใดๆ มีการรวบรวม ไลบรารีของ การถอดรหัส mRNA ของไหมและแปลงเป็นดีเอ็นเอเสริม (cDNA) เพื่อจัดเก็บโดยใช้เอนไซม์รีเวอร์สทรานสคริปเทสในช่วงปลายทศวรรษ 1970 [ 13 ]ในช่วงทศวรรษ 1980 มีการใช้การจัดลำดับแบบความเร็วต่ำโดยใช้ วิธี Sangerเพื่อจัดลำดับการถอดรหัสแบบสุ่ม ทำให้เกิดแท็กแสดงลำดับ (ESTs) [ 2 ] [ 14 ] [ 15 ] [ 16 ]วิธีการจัดลำดับแบบ Sangerเป็นที่แพร่หลายจนกระทั่งมีการคิดค้นวิธีการความเร็วสูงเช่นการจัดลำดับโดยการสังเคราะห์ (Solexa/Illumina) ESTsกลายเป็นที่นิยมในช่วงทศวรรษ 1990 ในฐานะวิธีการที่มีประสิทธิภาพในการกำหนดเนื้อหาของยีนของสิ่งมีชีวิตโดยไม่ต้องจัดลำดับจีโนมทั้งหมด[ 16 ]ปริมาณของทรานสคริปต์แต่ละรายการถูกวัดปริมาณโดยใช้Northern blotting , อาร์เรย์เมมเบรนไนลอนและต่อมา ใช้วิธี การ PCR เชิงปริมาณแบบ reverse transcriptase (RT-qPCR) [ 17 ] [ 18 ]แต่วิธีการเหล่านี้ยุ่งยากและสามารถจับได้เพียงส่วนย่อยเล็กๆ ของทรานสคริปโตมเท่านั้น[ 12 ]ด้วยเหตุนี้ วิธีการที่ทรานสคริปโตมทั้งหมดถูกแสดงออกและควบคุมจึงยังไม่เป็นที่ทราบแน่ชัดจนกระทั่งมีการพัฒนาเทคนิคที่มีประสิทธิภาพสูงขึ้น
ความพยายามในช่วงแรก
คำว่า "transcriptome" ถูกใช้ครั้งแรกในช่วงทศวรรษ 1990 [ 19 ] [ 20 ]ในปี 1995 หนึ่งในวิธีการทรานสคริปโตมิกส์ที่ใช้การจัดลำดับแบบแรกๆ ได้รับการพัฒนาขึ้น นั่นคือ การวิเคราะห์การแสดงออกของยีนแบบอนุกรม (SAGE) ซึ่งทำงานโดยการจัดลำดับแบบ Sangerของชิ้นส่วนทรานสคริปต์แบบสุ่มที่เชื่อมต่อกัน[ 21 ]ทรานสคริปต์จะถูกวัดปริมาณโดยการจับคู่ชิ้นส่วนกับยีนที่รู้จัก นอกจากนี้ยังมีการใช้ SAGE เวอร์ชันที่ใช้เทคนิคการจัดลำดับแบบความเร็วสูง เรียกว่า การวิเคราะห์การแสดงออกของยีนแบบดิจิทัล ในช่วงสั้นๆ[ 9 ] [ 22 ]อย่างไรก็ตาม วิธีการเหล่านี้ส่วนใหญ่ถูกแทนที่ด้วยการจัดลำดับแบบความเร็วสูงของทรานสคริปต์ทั้งหมด ซึ่งให้ข้อมูลเพิ่มเติมเกี่ยวกับโครงสร้างของทรานสคริปต์ เช่นตัวแปรการเชื่อมต่อ[ 9 ]
การพัฒนาเทคนิคสมัยใหม่
| RNA-Seq | ไมโครอาร์เรย์ | |
|---|---|---|
| อัตราการไหลผ่าน | 1 วันถึง 1 สัปดาห์ต่อการทดลอง[ 10 ] | 1–2 วันต่อการทดลอง[ 10 ] |
| ปริมาณ RNA ที่ป้อนเข้า | RNA ทั้งหมดต่ำ ~ 1 ng [ 25 ] | สูง ~ 1 μg mRNA [ 26 ] |
| ความเข้มข้นของแรงงาน | สูง (การเตรียมตัวอย่างและการวิเคราะห์ข้อมูล) [ 10 ] [ 23 ] | ต่ำ[ 10 ] [ 23 ] |
| ความรู้เดิม | ไม่จำเป็นต้องระบุ แต่ลำดับจีโนม/ทรานสคริปโตมอ้างอิงจะมีประโยชน์[ 23 ] | จีโนม/ทรานสคริปโตมอ้างอิงจำเป็นสำหรับการออกแบบโพรบ[ 23 ] |
| ความแม่นยำในการวัดปริมาณ | ~90% (จำกัดโดยความครอบคลุมของลำดับ) [ 27 ] | >90% (จำกัดโดยความแม่นยำในการตรวจจับฟลูออเรสเซนซ์) [ 27 ] |
| ความละเอียดของลำดับ | RNA-Seq สามารถตรวจจับSNPและ splice variant ได้ (จำกัดด้วยความแม่นยำในการจัดลำดับประมาณ 99%) [ 27 ] | อาร์เรย์เฉพาะทางสามารถตรวจจับตัวแปรการต่อเชื่อม mRNA ได้ (จำกัดโดยการออกแบบโพรบและการผสมข้าม) [ 27 ] |
| ความไว | 1 ทรานสคริปต์ต่อล้าน (โดยประมาณ จำกัดโดยความครอบคลุมของลำดับ) [ 27 ] | 1 ทรานสคริปต์ต่อพัน (โดยประมาณ จำกัดโดยการตรวจจับฟลูออเรสเซนซ์) [ 27 ] |
| ช่วงไดนามิก | 100,000:1 (จำกัดโดยความครอบคลุมของลำดับ) [ 28 ] | 1,000:1 (จำกัดโดยความอิ่มตัวของฟลูออเรสเซนซ์) [ 28 ] |
| ความสามารถในการทำซ้ำทางเทคนิค | >99% [ 29 ] [ 30 ] | >99% [ 31 ] [ 32 ] |
เทคนิคที่โดดเด่นในปัจจุบัน ได้แก่ไมโครอาร์เรย์และRNA-Seqได้รับการพัฒนาในช่วงกลางทศวรรษ 1990 และ 2000 [ 9 ] [ 33 ]ไมโครอาร์เรย์ที่วัดปริมาณของชุดทรานสคริปต์ที่กำหนดไว้ผ่านการไฮบริดกับอาร์เรย์ของโพรบเสริม ได้รับการตีพิมพ์ครั้งแรกในปี 1995 [ 34 ] [ 35 ]เทคโนโลยีไมโครอาร์เรย์ทำให้สามารถวิเคราะห์ทรานสคริปต์หลายพันรายการพร้อมกันได้ในราคาที่ลดลงอย่างมากต่อยีนและประหยัดแรงงาน[ 36 ]ทั้งอาร์เรย์โอลิโกนิวคลีโอไทด์แบบจุดและ อาร์เรย์ความหนาแน่นสูง ของ Affymetrixเป็นวิธีการที่เลือกใช้สำหรับการสร้างโปรไฟล์การถอดรหัสจนถึงปลายทศวรรษ 2000 [ 12 ] [ 33 ]ในช่วงเวลานี้ มีการผลิตไมโครอาร์เรย์หลากหลายชนิดเพื่อครอบคลุมยีนที่รู้จักใน สิ่งมีชีวิต แบบจำลองหรือสิ่งมีชีวิตที่มีความสำคัญทางเศรษฐกิจ ความก้าวหน้าในการออกแบบและการผลิตอาร์เรย์ช่วยปรับปรุงความจำเพาะของโพรบและทำให้สามารถทดสอบยีนได้มากขึ้นบนอาร์เรย์เดียว ความก้าวหน้าในการตรวจจับฟลูออเรสเซนซ์ทำให้ความไวและความแม่นยำในการวัดเพิ่มขึ้นสำหรับทรานสคริปต์ที่มีปริมาณน้อย[ 35 ] [ 37 ]
RNA-Seq ทำได้โดยการถอดรหัส RNA ย้อนกลับในหลอดทดลองและจัดลำดับ cDNA ที่ได้[ 10 ]ความอุดมสมบูรณ์ของทรานสคริปต์ได้มาจากจำนวนนับจากทรานสคริปต์แต่ละรายการ ดังนั้นเทคนิคนี้จึงได้รับอิทธิพลอย่างมากจากการพัฒนา เทคโนโลยีการจัดลำดับ แบบความเร็วสูง[ 9 ] [ 11 ]การจัดลำดับลายเซ็นแบบขนานขนาดใหญ่ (MPSS) เป็นตัวอย่างแรกๆ ที่ใช้การสร้างลำดับ 16–20 bpผ่านชุดไฮบริดไดเซชัน ที่ซับซ้อน [ 38 ] [หมายเหตุ 1 ] และถูกนำมาใช้ในปี 2547 เพื่อตรวจสอบการแสดงออกของยีนจำนวนหมื่นยีนในArabidopsis thaliana [ 39 ] งาน RNA-Seq ที่เก่าแก่ที่สุดได้รับการตีพิมพ์ในปี 2549 โดยมีการจัดลำดับทรานสคริปต์จำนวนหนึ่งแสนรายการโดยใช้เทคโนโลยี454 [ 40 ]ซึ่งมีความครอบคลุมเพียงพอที่จะวัดปริมาณความอุดมสมบูรณ์ของทรานสคริปต์สัมพัทธ์ RNA-Seq เริ่มได้รับความนิยมมากขึ้นหลังจากปี 2008 เมื่อเทคโนโลยี Solexa/Illumina ใหม่ ทำให้สามารถบันทึกลำดับทรานสคริปต์ได้ถึงหนึ่งพันล้านรายการ[ 4 ] [ 10 ] [ 41 ] [ 42 ]ผลผลิตนี้ทำให้สามารถวัดปริมาณและเปรียบเทียบทรานสคริปโตมของมนุษย์ ได้ [ 43 ]
การรวบรวมข้อมูล
การสร้างข้อมูลเกี่ยวกับทรานสคริปต์ RNA สามารถทำได้โดยใช้หลักการหลักสองประการ ได้แก่ การจัดลำดับทรานสคริปต์แต่ละรายการ ( ESTหรือ RNA-Seq) หรือการไฮบริดไดเซชันของทรานสคริปต์กับอาร์เรย์ของโพรบนิวคลีโอไทด์ที่เรียงลำดับ (ไมโครอาร์เรย์) [ 23 ]
การแยกอาร์เอ็นเอ
วิธีการทรานสคริปโตมิกทั้งหมดจำเป็นต้องแยก RNA ออกจากสิ่งมีชีวิตทดลองก่อนจึงจะสามารถบันทึกทรานสคริปต์ได้ แม้ว่าระบบชีวภาพจะมีความหลากหลายอย่างมาก แต่ เทคนิค การสกัด RNA นั้นโดยทั่วไปแล้วคล้ายคลึงกันและเกี่ยวข้องกับการทำลายเซลล์หรือเนื้อเยื่อด้วยกลไก การทำลายRNaseด้วยเกลือ chaotropic [ 44 ]การทำลายโมเลกุลขนาดใหญ่และสารประกอบนิวคลีโอไทด์ การแยก RNA ออกจากโมเลกุลชีวภาพที่ไม่ต้องการรวมถึง DNAและการเพิ่มความเข้มข้นของ RNA ผ่านการตกตะกอนจากสารละลายหรือการชะล้างจากเมทริกซ์ของแข็ง[ 44 ] [ 45 ]นอกจากนี้ RNA ที่แยกได้อาจได้รับการบำบัดด้วยDNaseเพื่อย่อย DNA ที่เหลืออยู่[ 46 ]จำเป็นต้องเพิ่มความเข้มข้นของ messenger RNA เนื่องจากสารสกัด RNA ทั้งหมดโดยทั่วไปมีribosomal RNAถึง 98% [ 47 ]การเพิ่มความเข้มข้นของทรานสคริปต์สามารถทำได้โดย วิธีการจับกับ poly-Aหรือโดยการลดปริมาณ ribosomal RNA โดยใช้โพรบที่จำเพาะต่อลำดับ[ 48 ] RNA ที่เสื่อมสภาพอาจส่งผลต่อผลลัพธ์ในขั้นตอนต่อไป ตัวอย่างเช่น การเพิ่มความเข้มข้นของ mRNA จากตัวอย่างที่เสื่อมสภาพจะส่งผลให้ปลาย5' ของ mRNA ลด ลง และสัญญาณไม่สม่ำเสมอตลอดความยาวของทรานสคริปต์การแช่แข็งเนื้อเยื่ออย่างรวดเร็วก่อนการแยก RNA เป็นเรื่องปกติ และต้องระมัดระวังเพื่อลดการสัมผัสกับเอนไซม์ RNase เมื่อการแยกเสร็จสมบูรณ์[ 45 ]
แท็กลำดับที่แสดงออก
แท็กแสดงลำดับ (EST) คือลำดับนิวคลีโอไทด์สั้นๆ ที่สร้างขึ้นจากทรานสคริปต์ RNA เดียว RNA จะถูกคัดลอกเป็นDNA เสริม (cDNA) โดย เอนไซม์ รีเวอร์สทรานสคริปเทสก่อน จากนั้นจึงนำ cDNA ที่ได้ไปจัดลำดับ[ 16 ] เนื่องจากสามารถรวบรวม EST ได้โดยไม่ต้องมีความรู้มาก่อนเกี่ยวกับสิ่งมีชีวิตที่มาจาก EST จึงสามารถสร้างได้จากส่วนผสมของสิ่งมีชีวิตหรือตัวอย่างสิ่งแวดล้อม[ 49 ] [ 16 ]แม้ว่าปัจจุบันจะใช้วิธีการที่มีปริมาณงานสูงกว่า แต่ โดยทั่วไปแล้ว ไลบรารี ESTให้ข้อมูลลำดับสำหรับการออกแบบไมโครอาร์เรย์ในยุคแรกๆ ตัวอย่างเช่น ไมโครอาร์เรย์ ข้าวบาร์เลย์ได้รับการออกแบบจาก EST ที่จัดลำดับไว้ก่อนหน้านี้ 350,000 รายการ[ 50 ]
การวิเคราะห์การแสดงออกของยีนแบบอนุกรมและแบบแคป (SAGE/CAGE)

การวิเคราะห์การแสดงออกของยีนแบบอนุกรม (SAGE) เป็นการพัฒนาวิธีการ EST เพื่อเพิ่มปริมาณแท็กที่สร้างขึ้นและอนุญาตให้มีการวัดปริมาณความอุดมสมบูรณ์ของทรานสคริปต์ได้[ 21 ] cDNAถูกสร้างขึ้นจากRNAแต่จะถูกย่อยเป็น ชิ้นส่วน "แท็ก" ขนาด 11 bp โดยใช้เอนไซม์จำกัดที่ตัด DNA ที่ลำดับเฉพาะ และ 11 คู่เบสถัดจากลำดับนั้น แท็ก cDNA เหล่านี้จะถูกต่อกันแบบหัวต่อท้ายเป็นสายยาว (>500 bp) และจัดลำดับโดยใช้วิธีการที่มีปริมาณงานต่ำ แต่มีความยาวในการอ่านสูง เช่นการจัดลำดับแบบ Sangerจากนั้นลำดับจะถูกแบ่งกลับเป็นแท็ก 11 bp ดั้งเดิมโดยใช้ซอฟต์แวร์คอมพิวเตอร์ในกระบวนการที่เรียกว่าการแยกส่วน [ 21 ] หาก มี จีโนมอ้างอิงคุณภาพสูงแท็กเหล่านี้อาจจับคู่กับยีนที่สอดคล้องกันในจีโนมได้ หากไม่มีจีโนมอ้างอิง แท็กเหล่านี้สามารถใช้เป็นเครื่องหมายวินิจฉัยได้โดยตรงหากพบว่ามีการแสดงออกที่แตกต่างกันในสภาวะของโรค[ 21 ]
วิธีการวิเคราะห์การแสดงออกของยีนด้วยแคป (CAGE) เป็นรูปแบบหนึ่งของ SAGE ที่จัดลำดับแท็กจากปลาย 5'ของทรานสคริปต์ mRNA เท่านั้น[ 52 ]ดังนั้นตำแหน่งเริ่มต้นการถอดรหัสของยีนสามารถระบุได้เมื่อแท็กถูกจัดเรียงกับจีโนมอ้างอิง การระบุตำแหน่งเริ่มต้นของยีนมีประโยชน์สำหรับ การวิเคราะห์ โปรโมเตอร์และการโคลน cDNA แบบเต็มความยาว
วิธีการ SAGE และ CAGE ให้ข้อมูลเกี่ยวกับยีนมากกว่าที่เคยเป็นไปได้เมื่อทำการลำดับ EST เดี่ยว แต่การเตรียมตัวอย่างและการวิเคราะห์ข้อมูลมักจะใช้แรงงานมากกว่า[ 52 ]
ไมโครอาร์เรย์
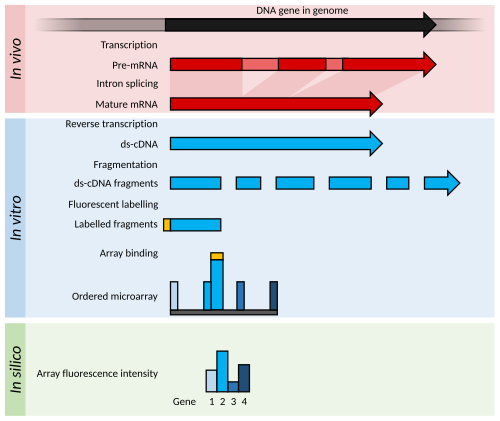
หลักการและความก้าวหน้า
ไมโครอาร์เรย์มักประกอบด้วยตารางของโอลิโกเมอร์นิว คลีโอไทด์สั้นๆ ที่เรียกว่า " โพรบ " ซึ่งโดยทั่วไปจะจัดเรียงอยู่บนแผ่นกระจก[ 53 ]ปริมาณทรานสคริปต์จะถูกกำหนดโดยการไฮบริดไดเซชันของ ทรานสคริปต์ที่ติดฉลาก ด้วยฟลูออเรสเซนต์กับโพรบเหล่านี้[ 54 ]ความเข้มของฟลูออเรสเซนต์ที่ตำแหน่งโพรบแต่ละตำแหน่งบนอาร์เรย์จะบ่งชี้ถึงปริมาณทรานสคริปต์สำหรับลำดับโพรบนั้น[ 54 ]กลุ่มของโพรบที่ออกแบบมาเพื่อวัดทรานสคริปต์เดียวกัน (เช่น การไฮบริดไดเซชันทรานสคริปต์เฉพาะในตำแหน่งต่างๆ) มักเรียกว่า "ชุดโพรบ"
ไมโครอาร์เรย์ต้องการความรู้ทางจีโนมบางส่วนจากสิ่งมีชีวิตที่สนใจ เช่น ในรูปแบบของ ลำดับ จีโนมที่มีคำอธิบายประกอบ หรือไลบรารีของ EST ที่สามารถใช้สร้างโพรบสำหรับอาร์เรย์ได้[ 36 ]
วิธีการ
โดยทั่วไปไมโครอาร์เรย์สำหรับทรานสคริปโตมิกส์จะแบ่งออกเป็นสองประเภทใหญ่ๆ คือ อาร์เรย์จุดความหนาแน่นต่ำ หรืออาร์เรย์โพรบสั้นความหนาแน่นสูง ปริมาณทรานสคริปต์จะถูกอนุมานจากความเข้มของฟลูออเรสเซนซ์ที่ได้จากทรานสคริปต์ที่ติดแท็กฟลูออโรฟอร์ซึ่งจับกับอาร์เรย์[ 36 ]
โดยทั่วไปแล้ว อาร์เรย์ความหนาแน่นต่ำแบบจุดจะมี หยด cDNAบริสุทธิ์หลากหลายชนิดในปริมาณพิโคลิตร[หมายเหตุ 2 ]เรียงตัวอยู่บนพื้นผิวของแผ่นกระจก[ 55 ]โพรบเหล่านี้มีความยาวมากกว่าโพรบของอาร์เรย์ความหนาแน่นสูงและไม่สามารถระบุ เหตุการณ์ การตัดต่อทางเลือกได้อาร์เรย์แบบจุดใช้ฟลูออโรฟอร์ สองชนิดที่แตกต่างกัน เพื่อติดฉลากตัวอย่างทดสอบและตัวอย่างควบคุม และอัตราส่วนของฟลูออเรสเซนซ์จะถูกใช้ในการคำนวณปริมาณสัมพัทธ์[ 56 ]อาร์เรย์ความหนาแน่นสูงใช้ฉลากฟลูออเรสเซนซ์เพียงชนิดเดียว และแต่ละตัวอย่างจะถูกไฮบริดและตรวจจับทีละตัว[ 57 ]อาร์เรย์ความหนาแน่นสูงได้รับความนิยมจาก อาร์เรย์ Affymetrix GeneChipซึ่งแต่ละทรานสคริปต์จะถูกวัดปริมาณโดยโพรบ 25 -mer สั้นๆ หลายตัว ที่ร่วมกันตรวจสอบยีนหนึ่งตัว[ 58 ]
อาร์เรย์ NimbleGen เป็นอาร์เรย์ความหนาแน่นสูงที่ผลิตขึ้นโดย วิธี โฟโตเคมีแบบไม่มีหน้ากากซึ่งช่วยให้สามารถผลิตอาร์เรย์ได้อย่างยืดหยุ่นในจำนวนน้อยหรือมาก อาร์เรย์เหล่านี้มีโพรบ 45 ถึง 85 เมอร์หลายแสนตัว และถูกไฮบริดกับตัวอย่างที่ติดฉลากสีเดียวสำหรับการวิเคราะห์การแสดงออก[ 59 ]บางแบบมีการรวมอาร์เรย์อิสระมากถึง 12 อาร์เรย์ต่อสไลด์
RNA-Seq

หลักการและความก้าวหน้า
RNA-Seqหมายถึงการผสมผสานระหว่าง วิธี การจัดลำดับแบบความเร็วสูงกับวิธีการคำนวณเพื่อจับภาพและวัดปริมาณทรานสคริปต์ที่มีอยู่ในสารสกัด RNA [ 10 ]ลำดับนิวคลีโอไทด์ที่สร้างขึ้นโดยทั่วไปมีความยาวประมาณ 100 bp แต่สามารถมีช่วงตั้งแต่ 30 bp ถึงมากกว่า 10,000 bp ขึ้นอยู่กับวิธีการจัดลำดับที่ใช้ RNA-Seq ใช้ประโยชน์จาก การสุ่มตัวอย่างท รานสคริปโตมอย่างละเอียดด้วยชิ้นส่วนสั้นๆ จำนวนมากจากทรานสคริปโตมเพื่อให้สามารถสร้างทรานสคริปต์ RNA ดั้งเดิมขึ้นใหม่ได้ด้วยวิธีการคำนวณโดยการจัดเรียงลำดับการอ่านกับจีโนมอ้างอิงหรือกับกันและกัน ( การประกอบแบบ de novo ) [ 9 ]ทั้ง RNA ที่มีปริมาณน้อยและปริมาณมากสามารถวัดปริมาณได้ในการทดลอง RNA-Seq ( ช่วงไดนามิก 5 ลำดับความ magnitude ) ซึ่งเป็นข้อได้เปรียบที่สำคัญเหนือทรานสคริปโตมของไมโครอาร์เรย์ นอกจากนี้ ปริมาณ RNA ที่ใช้ในการป้อนข้อมูลสำหรับ RNA-Seq (ปริมาณระดับนาโนกรัม) ยังต่ำกว่ามากเมื่อเทียบกับไมโครอาร์เรย์ (ปริมาณระดับไมโครกรัม) ซึ่งช่วยให้สามารถตรวจสอบทรานสคริปโตมได้แม้ในระดับความละเอียดของเซลล์เดียวเมื่อรวมกับการขยาย cDNA [ 25 ] [ 60 ]ในทางทฤษฎีแล้ว ไม่มีขีดจำกัดสูงสุดของการหาปริมาณใน RNA-Seq และสัญญาณรบกวนพื้นหลังต่ำมากสำหรับการอ่าน 100 bp ในบริเวณที่ไม่ซ้ำกัน[ 10 ]
RNA-Seq อาจใช้เพื่อระบุยีนภายในจีโนมหรือระบุว่ายีนใดทำงาน ณ จุดเวลาใดเวลาหนึ่ง และจำนวนการอ่านสามารถใช้เพื่อสร้างแบบจำลองระดับการแสดงออกของยีนสัมพัทธ์ได้อย่างแม่นยำ วิธีการ RNA-Seq ได้รับการปรับปรุงอย่างต่อเนื่อง โดยส่วนใหญ่ผ่านการพัฒนาเทคโนโลยีการจัดลำดับดีเอ็นเอเพื่อเพิ่มปริมาณงาน ความแม่นยำ และความยาวของการอ่าน[ 61 ]นับตั้งแต่คำอธิบายครั้งแรกในปี 2549 และ 2551 [ 40 ] [ 62 ] RNA-Seq ได้รับการนำไปใช้อย่างรวดเร็วและแซงหน้าไมโครอาร์เรย์ในฐานะเทคนิคทรานสคริปโตมิกส์ที่โดดเด่นในปี 2558 [ 63 ]
การแสวงหาข้อมูลทรานสคริปโตมในระดับเซลล์แต่ละเซลล์ได้ผลักดันให้เกิดความก้าวหน้าในวิธีการเตรียมไลบรารี RNA-Seq ส่งผลให้ความไวเพิ่มขึ้นอย่างมาก ปัจจุบัน ทรานสคริปโตมของเซลล์เดี่ยวได้รับการอธิบายอย่างละเอียดแล้ว และยังขยายไปถึง RNA-Seq ใน แหล่งกำเนิดซึ่งทรานสคริปโตมของเซลล์แต่ละเซลล์จะถูกตรวจสอบโดยตรงในเนื้อเยื่อที่ตรึงไว้[ 64 ]
วิธีการ
RNA-Seq ถูกสร้างขึ้นควบคู่ไปกับการพัฒนาอย่างรวดเร็วของเทคโนโลยีการจัดลำดับดีเอ็นเอที่มีปริมาณมาก[ 65 ]อย่างไรก็ตาม ก่อนที่จะมีการจัดลำดับทรานสคริปต์ RNA ที่สกัดออกมา จะมีการดำเนินการขั้นตอนการประมวลผลที่สำคัญหลายขั้นตอน วิธีการจะแตกต่างกันในการใช้การเพิ่มความเข้มข้นของทรานสคริปต์ การแตกตัว การขยาย การจัดลำดับแบบปลายเดี่ยวหรือแบบคู่ และว่าจะรักษาข้อมูลสายไว้หรือไม่[ 65 ]
ความไวของการทดลอง RNA-Seq สามารถเพิ่มขึ้นได้โดยการเพิ่มความเข้มข้นของ RNA ที่น่าสนใจและลด RNA ที่มีอยู่มากที่ทราบแล้ว โมเลกุล mRNA สามารถแยกได้โดยใช้โพรบโอลิโกนิวคลีโอไทด์ที่จับกับหางโพลี-A ของพวกมัน หรืออีกทางหนึ่ง สามารถใช้การกำจัดไรโบโซมเพื่อกำจัดRNA ไรโบโซม (rRNA) ที่มีอยู่มากแต่ไม่มีข้อมูลโดยเฉพาะ โดยการไฮบริดกับโพรบที่ปรับให้เข้ากับลำดับ rRNA เฉพาะของกลุ่มสิ่งมีชีวิต (เช่น rRNA ของสัตว์เลี้ยงลูกด้วยนม , rRNA ของพืช) อย่างไรก็ตาม การกำจัดไรโบโซมอาจทำให้เกิดอคติบางอย่างผ่านการกำจัดทรานสคริปต์นอกเป้าหมายที่ไม่เฉพาะเจาะจง[ 66 ] RNA ขนาดเล็ก เช่นไมโคร RNAสามารถทำให้บริสุทธิ์ตามขนาดได้โดยการ อิ เล็กโทรโฟเรซิสแบบเจลและการสกัด
เนื่องจาก mRNA มีความยาวมากกว่าความยาวของการอ่านของวิธีการจัดลำดับแบบความเร็วสูงทั่วไป จึงมักจะต้องแบ่งทรานสคริปต์ออกเป็นส่วนย่อยก่อนการจัดลำดับ[ 67 ]วิธีการแบ่งส่วนย่อยเป็นประเด็นสำคัญในการสร้างไลบรารีการจัดลำดับการแบ่งส่วนย่อยอาจทำได้โดยการไฮโดรไลซิสทางเคมีการพ่นละออง การใช้ คลื่นเสียงหรือการถอดรหัสย้อนกลับด้วย นิวคลีโอไท ด์ที่หยุดสายโซ่[ 67 ]หรืออีกทางหนึ่ง การแบ่งส่วนย่อยและการติดแท็ก cDNA อาจทำพร้อมกันโดยใช้เอนไซม์ทรานสโพเซส[ 68 ]
ในระหว่างการเตรียมการลำดับดีเอ็นเอ สำเนา cDNA ของทรานสคริปต์อาจถูกขยายด้วยPCRเพื่อเพิ่มความเข้มข้นของชิ้นส่วนที่มีลำดับอะแดปเตอร์ 5' และ 3' ที่คาดไว้[ 69 ]การขยายยังใช้เพื่อให้สามารถลำดับ RNA ในปริมาณอินพุตที่ต่ำมาก ลงไปถึง 50 pgในการใช้งานที่รุนแรง[ 70 ]ตัวควบคุมสไปค์อินของ RNA ที่ทราบแล้วสามารถใช้สำหรับการประเมินการควบคุมคุณภาพเพื่อตรวจสอบการเตรียมไลบรารีและการลำดับ ในแง่ของปริมาณ GCความยาวของชิ้นส่วน ตลอดจนอคติเนื่องจากตำแหน่งของชิ้นส่วนภายในทรานสคริปต์[ 71 ]ตัวระบุโมเลกุลที่ไม่ซ้ำกัน (UMIs) คือลำดับสุ่มสั้นๆ ที่ใช้ในการติดแท็กชิ้นส่วนลำดับแต่ละชิ้นในระหว่างการเตรียมไลบรารี เพื่อให้ชิ้นส่วนที่ติดแท็กแต่ละชิ้นมีเอกลักษณ์เฉพาะตัว[ 72 ] UMIs ให้มาตราส่วนสัมบูรณ์สำหรับการหาปริมาณ โอกาสในการแก้ไขอคติการขยายที่เกิดขึ้นภายหลังในระหว่างการสร้างไลบรารี และประมาณขนาดตัวอย่างเริ่มต้นได้อย่างแม่นยำ UMI เหมาะอย่างยิ่งสำหรับทรานสคริปโตมิกส์ RNA-Seq เซลล์เดี่ยว ซึ่งปริมาณ RNA ขาเข้ามีจำกัดและจำเป็นต้องมีการขยายตัวอย่างอย่างต่อเนื่อง[ 73 ] [ 74 ] [ 75 ]
เมื่อเตรียมโมเลกุลของทรานสคริปต์เสร็จแล้ว ก็สามารถจัดลำดับได้ในทิศทางเดียว (single-end) หรือทั้งสองทิศทาง (paired-end) การจัดลำดับแบบ single-end มักจะทำได้เร็วกว่า ราคาถูกกว่าการจัดลำดับแบบ paired-end และเพียงพอสำหรับการหาปริมาณระดับการแสดงออกของยีน การจัดลำดับแบบ paired-end จะสร้างการจัดเรียง/การประกอบที่แข็งแกร่งกว่า ซึ่งเป็นประโยชน์สำหรับการระบุยีนและการค้นพบไอโซฟอร์ม ของทรานสคริปต์ [ 10 ]วิธีการ RNA-Seq ที่เฉพาะเจาะจงสายจะรักษา ข้อมูล สายของทรานสคริปต์ที่จัดลำดับไว้[ 76 ]หากไม่มีข้อมูลสาย การอ่านสามารถจัดเรียงกับตำแหน่งยีน ได้ แต่จะไม่บอกว่ายีนนั้นถูกถอดรหัสในทิศทางใด Stranded-RNA-Seq มีประโยชน์สำหรับการถอดรหัสการถอดรหัสสำหรับยีนที่ทับซ้อนกันในทิศทางต่างๆ และเพื่อทำนายยีนได้อย่างแม่นยำยิ่งขึ้นในสิ่งมีชีวิตที่ไม่ใช่แบบจำลอง[ 76 ]
| แพลตฟอร์ม | การวางจำหน่ายเชิงพาณิชย์ | ความยาวในการอ่านโดยทั่วไป | ปริมาณงานสูงสุดต่อรอบ | ความแม่นยำในการอ่านครั้งเดียว | ผลการวิเคราะห์ RNA-Seq ที่ฝากไว้ใน NCBI SRA (ตุลาคม 2016) [ 79 ] |
|---|---|---|---|---|---|
| 454 วิทยาศาสตร์ชีวภาพ | 2548 | 700 bp | 0.7 ปอนด์ | 99.9% | 3548 |
| อิลลูมิน่า | 2006 | 50–300 bp | 900 ปอนด์ | 99.9% | 362903 |
| แข็ง | 2008 | 50 บีพี | 320 ปอนด์ | 99.9% | 7032 |
| ไอออนทอร์เรนต์ | 2010 | 400 bp | 30 ปอนด์ | 98% | 1953 |
| แพคไบโอ | 2011 | 10,000 บีพี | 2 ปอนด์ | 87% | 160 |
คำอธิบายสัญลักษณ์: NCBI SRA – ศูนย์ข้อมูลเทคโนโลยีชีวภาพแห่งชาติ (National Center for Biotechnology Information) คลังเก็บข้อมูลลำดับนิวคลีโอไทด์ (Sequence Read Archive)
ปัจจุบัน RNA-Seq อาศัยการคัดลอกโมเลกุล RNA ไปเป็นโมเลกุล cDNA ก่อนการจัดลำดับ ดังนั้นแพลตฟอร์มที่ใช้จึงเหมือนกันสำหรับข้อมูลทรานสคริปโตมิกและจีโนมิก ด้วยเหตุนี้ การพัฒนาเทคโนโลยีการจัดลำดับ DNA จึงเป็นคุณลักษณะสำคัญของ RNA-Seq [ 78 ] [ 80 ] [ 81 ]การจัดลำดับ RNA โดยตรงโดยใช้การจัดลำดับนาโนพอเรถือเป็นเทคนิค RNA-Seq ที่ทันสมัยที่สุดในปัจจุบัน[ 82 ] [ 83 ]การจัดลำดับ RNA ด้วยนาโนพอเรสามารถตรวจจับเบสที่ดัดแปลงซึ่งอาจถูกบดบังเมื่อจัดลำดับ cDNA และยังช่วยขจัด ขั้นตอน การขยายสัญญาณที่อาจทำให้เกิดความลำเอียงได้[ 11 ] [ 84 ]
ความไวและความแม่นยำของการทดลอง RNA-Seq ขึ้นอยู่กับจำนวนการอ่านที่ได้รับจากแต่ละตัวอย่าง[ 85 ] [ 86 ]จำเป็นต้องมีการอ่านจำนวนมากเพื่อให้แน่ใจว่าครอบคลุมทรานสคริปโตมอย่างเพียงพอ ทำให้สามารถตรวจจับทรานสคริปต์ที่มีความอุดมสมบูรณ์ต่ำได้ การออกแบบการทดลองมีความซับซ้อนมากขึ้นด้วยเทคโนโลยีการจัดลำดับที่มีช่วงเอาต์พุตจำกัด ประสิทธิภาพการสร้างลำดับที่แปรผัน และคุณภาพลำดับที่แปรผัน นอกจากนี้ยังต้องพิจารณาด้วยว่าแต่ละสปีชีส์มีจำนวนยีน ที่แตกต่างกัน ดังนั้นจึงต้องการผลผลิตลำดับที่ปรับแต่งให้เหมาะสมสำหรับทรานสคริปโตมที่มีประสิทธิภาพ การศึกษาในช่วงแรกกำหนดเกณฑ์ที่เหมาะสมโดยอาศัยประสบการณ์ แต่เมื่อเทคโนโลยีพัฒนาขึ้น การครอบคลุมที่เหมาะสมจะถูกทำนายโดยการคำนวณจากการอิ่มตัวของทรานสคริปโตม วิธีที่มีประสิทธิภาพที่สุดในการปรับปรุงการตรวจจับการแสดงออกที่แตกต่างกันในยีนที่มีการแสดงออกต่ำคือการเพิ่มตัวอย่างทางชีวภาพมากกว่าการเพิ่มการอ่าน[ 87 ]เกณฑ์มาตรฐานปัจจุบันที่แนะนำโดย โครงการ Encyclopedia of DNA Elements (ENCODE) คือการครอบคลุมเอ็กโซม 70 เท่าสำหรับ RNA-Seq มาตรฐาน และการครอบคลุมเอ็กโซมสูงสุด 500 เท่าเพื่อตรวจจับทรานสคริปต์และไอโซฟอร์มที่หายาก[ 88 ] [ 89 ] [ 90 ]
การวิเคราะห์ข้อมูล
วิธีการทรานสคริปโตมิกส์เป็นแบบขนานสูงและต้องใช้การคำนวณจำนวนมากเพื่อสร้างข้อมูลที่มีความหมายสำหรับการทดลองไมโครอาร์เรย์และ RNA-Seq [ 91 ] [ 92 ] [ 93 ] [ 94 ] [ 95 ]ข้อมูลไมโครอาร์เรย์ถูกบันทึกเป็นภาพความละเอียดสูง ซึ่งต้องใช้ การตรวจจับคุณลักษณะและการวิเคราะห์สเปกตรัม[ 96 ]ไฟล์ภาพดิบของไมโครอาร์เรย์แต่ละไฟล์มีขนาดประมาณ 750 MB ในขณะที่ความเข้มที่ประมวลผลแล้วมีขนาดประมาณ 60 MB โพรบสั้นหลายตัวที่ตรงกับทรานสคริปต์เดียวสามารถเปิดเผยรายละเอียดเกี่ยวกับ โครงสร้าง อินตรอน - เอ็กซอนซึ่งต้องใช้แบบจำลองทางสถิติเพื่อกำหนดความถูกต้องของสัญญาณที่ได้ การศึกษา RNA-Seq สร้างลำดับ DNA สั้น ๆ นับพันล้านลำดับ ซึ่งต้องจัดเรียงให้ตรงกับจีโนมอ้างอิงที่ประกอบด้วยเบสคู่หลายล้านถึงหลายพันล้านคู่การประกอบอ่าน แบบ de novoภายในชุดข้อมูลต้องสร้างกราฟลำดับที่ ซับซ้อนมาก [ 97 ]การดำเนินการ RNA-Seq มีลักษณะซ้ำซ้อนสูงและได้รับประโยชน์จากการคำนวณแบบขนานแต่ด้วยอัลกอริทึมที่ทันสมัย ฮาร์ดแวร์คอมพิวเตอร์สำหรับผู้บริโภคก็เพียงพอสำหรับการทดลองทรานสคริปโตมิกส์แบบง่ายๆ ที่ไม่ต้องการการประกอบอ่านแบบde novo [ 98 ]สามารถบันทึกทรานสคริปโตมของมนุษย์ได้อย่างแม่นยำโดยใช้ RNA-Seq ด้วยลำดับ 100 bp จำนวน 30 ล้านลำดับต่อตัวอย่าง[ 85 ] [ 86 ]ตัวอย่างนี้จะต้องใช้พื้นที่ดิสก์ประมาณ 1.8 กิกะไบต์ต่อตัวอย่างเมื่อจัดเก็บในรูปแบบ fastq ที่บีบอัด ข้อมูลการนับที่ประมวลผลแล้วสำหรับแต่ละยีนจะมีขนาดเล็กกว่ามาก เทียบเท่ากับความเข้มของไมโครอาร์เรย์ที่ประมวลผลแล้ว ข้อมูลลำดับอาจถูกจัดเก็บไว้ในที่เก็บข้อมูลสาธารณะ เช่นSequence Read Archive (SRA) [ 99 ]ชุดข้อมูล RNA-Seq สามารถอัปโหลดผ่าน Gene Expression Omnibus ได้[ 100 ]
การประมวลผลภาพ

การประมวลผลภาพไมโครอาร์เรย์ต้องระบุตารางปกติของคุณลักษณะภายในภาพได้อย่างถูกต้อง และวัดปริมาณความเข้ม ของการเรืองแสง สำหรับแต่ละคุณลักษณะ อย่างอิสระ นอกจากนี้ยังต้องระบุและกำจัด สิ่งแปลกปลอมในภาพออกจากการวิเคราะห์โดยรวม ความเข้มของการเรืองแสงบ่งชี้ถึงความอุดมสมบูรณ์ของแต่ละลำดับโดยตรง เนื่องจากลำดับของโพรบแต่ละตัวบนอาร์เรย์เป็นที่ทราบอยู่แล้ว[ 102 ]
ขั้นตอนแรกของ RNA-seq ยังรวมถึงการประมวลผลภาพที่คล้ายคลึงกัน อย่างไรก็ตาม การแปลงภาพเป็นข้อมูลลำดับมักจะดำเนินการโดยอัตโนมัติโดยซอฟต์แวร์ของเครื่องมือ วิธีการจัดลำดับแบบสังเคราะห์ของ Illumina ส่งผลให้เกิดคลัสเตอร์จำนวนมากที่กระจายอยู่ทั่วพื้นผิวของเซลล์ไหล[ 103 ]เซลล์ไหลจะถูกถ่ายภาพมากถึงสี่ครั้งในระหว่างรอบการจัดลำดับแต่ละครั้ง โดยมีรอบทั้งหมดหลายสิบถึงหลายร้อยรอบ คลัสเตอร์ของเซลล์ไหลนั้นคล้ายคลึงกับจุดไมโครอาร์เรย์และต้องได้รับการระบุอย่างถูกต้องในช่วงเริ่มต้นของกระบวนการจัดลำดับ ใน วิธีการไพโรซี เควนซิ ง ของRocheความเข้มของแสงที่ปล่อยออกมาจะเป็นตัวกำหนดจำนวนนิวคลีโอไทด์ที่ต่อเนื่องกันในการทำซ้ำโฮโมพอลิเมอร์ มีวิธีการเหล่านี้หลายแบบ แต่ละแบบมีโปรไฟล์ข้อผิดพลาดที่แตกต่างกันสำหรับข้อมูลที่ได้[ 104 ]
การวิเคราะห์ข้อมูล RNA-Seq
การทดลอง RNA-Seq สร้างข้อมูลลำดับดิบจำนวนมากซึ่งต้องได้รับการประมวลผลเพื่อให้ได้ข้อมูลที่เป็นประโยชน์ การวิเคราะห์ข้อมูลมักต้องใช้ เครื่องมือ ซอฟต์แวร์ชีวสารสนเทศ หลายอย่างร่วมกัน (ดูเพิ่มเติมที่รายชื่อเครื่องมือชีวสารสนเทศ RNA-Seq ) ซึ่งแตกต่างกันไปตามการออกแบบและเป้าหมายของการทดลอง กระบวนการนี้สามารถแบ่งออกเป็นสี่ขั้นตอน ได้แก่ การควบคุมคุณภาพ การจัดเรียง การหาปริมาณ และการแสดงออกที่แตกต่างกัน[ 105 ]โปรแกรม RNA-Seq ที่ได้รับความนิยมส่วนใหญ่จะทำงานจากอินเทอร์เฟซบรรทัดคำสั่งไม่ว่าจะเป็นใน สภาพแวดล้อม Unixหรือภายในสภาพแวดล้อมทางสถิติR / Bioconductor [ 94 ]
การควบคุมคุณภาพ
ลำดับการอ่านไม่สมบูรณ์แบบ ดังนั้นความแม่นยำของแต่ละเบสในลำดับจึงจำเป็นต้องได้รับการประเมินสำหรับการวิเคราะห์ในขั้นตอนต่อไป ข้อมูลดิบจะถูกตรวจสอบเพื่อให้แน่ใจว่า: คะแนนคุณภาพสำหรับการเรียกเบสสูง ปริมาณ GC ตรงกับการกระจายที่คาดไว้ โมทีฟลำดับสั้น ( k-mers ) ไม่ปรากฏมากเกินไป และอัตราการทำซ้ำของการอ่านอยู่ในระดับที่ยอมรับได้[ 86 ]มีซอฟต์แวร์หลายตัวเลือกสำหรับการวิเคราะห์คุณภาพลำดับ รวมถึง FastQC และ FaQCs [ 106 ] [ 107 ]ความผิดปกติอาจถูกลบออก (การตัดแต่ง) หรือติดแท็กเพื่อการจัดการพิเศษในระหว่างกระบวนการในภายหลัง
การจัดแนว
เพื่อเชื่อมโยงความอุดมสมบูรณ์ของลำดับการอ่านกับการแสดงออกของยีนเฉพาะ ลำดับการถอดรหัสจะถูกจัดเรียงกับจีโนมอ้างอิงหรือจัดเรียงใหม่หากไม่มีจีโนมอ้างอิง[ 108 ] [ 109 ] [ 110 ]ความท้าทายที่สำคัญสำหรับซอฟต์แวร์การจัดเรียงลำดับได้แก่ ความเร็วที่เพียงพอที่จะอนุญาตให้จัดเรียงลำดับสั้น ๆ หลายพันล้านลำดับภายในกรอบเวลาที่เหมาะสม ความยืดหยุ่นในการรับรู้และจัดการกับการตัดต่ออินทรอนของ mRNA ยูคาริโอต และการกำหนดการอ่านที่แมปไปยังหลายตำแหน่งอย่างถูกต้อง ความก้าวหน้าของซอฟต์แวร์ได้แก้ไขปัญหาเหล่านี้อย่างมาก และความยาวของการอ่านลำดับที่เพิ่มขึ้นช่วยลดโอกาสของการจัดเรียงลำดับที่ไม่ชัดเจน รายชื่อของตัวจัดเรียงลำดับที่มีปริมาณงานสูงที่มีอยู่ในปัจจุบันได้รับการดูแลโดยEBI [ 111 ] [ 112 ]
การจัดเรียงลำดับ mRNA ของ ทราน สคริปต์หลักที่ได้จากยูคาริโอตให้ตรงกับจีโนมอ้างอิงนั้น จำเป็นต้องมีการจัดการ ลำดับ อินทรอน แบบพิเศษ ซึ่งไม่มีอยู่ใน mRNA ที่เจริญเต็มที่แล้ว[ 113 ]โปรแกรมจัดเรียงลำดับแบบอ่านสั้นจะทำการจัดเรียงเพิ่มเติมอีกรอบหนึ่ง ซึ่งออกแบบมาโดยเฉพาะเพื่อระบุจุดเชื่อมต่อการต่อเชื่อมโดยอาศัยลำดับไซต์การต่อเชื่อมแบบมาตรฐานและข้อมูลไซต์การต่อเชื่อมของอินทรอนที่ทราบ การระบุจุดเชื่อมต่อการต่อเชื่อมของอินทรอนจะป้องกันไม่ให้ลำดับอ่านถูกจัดเรียงผิดพลาดข้ามจุดเชื่อมต่อการต่อเชื่อม หรือถูกทิ้งไปโดยไม่ถูกต้อง ทำให้สามารถจัดเรียงลำดับอ่านไปยังจีโนมอ้างอิงได้มากขึ้น และปรับปรุงความแม่นยำของการประมาณการแสดงออกของยีน เนื่องจาก อาจมี การควบคุมยีนใน ระดับ ไอโซฟอร์ม mRNAการจัดเรียงที่คำนึงถึงการต่อเชื่อมยังช่วยให้สามารถตรวจจับการเปลี่ยนแปลงความอุดมสมบูรณ์ของไอโซฟอร์ม ซึ่งอาจสูญหายไปในการวิเคราะห์แบบรวม[ 114 ]
การประกอบแบบ de novoสามารถใช้ในการจัดเรียงลำดับการอ่านเข้าด้วยกันเพื่อสร้างลำดับทรานสคริปต์แบบเต็มความยาวโดยไม่ต้องใช้จีโนมอ้างอิง[ 115 ]ความท้าทายเฉพาะของ การประกอบแบบ de novoได้แก่ ความต้องการการคำนวณที่มากกว่าเมื่อเทียบกับทรานสคริปโตมที่อิงตามอ้างอิง การตรวจสอบความถูกต้องเพิ่มเติมของตัวแปรยีนหรือชิ้นส่วน และคำอธิบายประกอบเพิ่มเติมของทรานสคริปต์ที่ประกอบแล้ว เมตริกแรกที่ใช้ในการอธิบายการประกอบทรานสคริปโตม เช่นN50ได้รับการพิสูจน์แล้วว่าทำให้เข้าใจผิด[ 116 ]และขณะนี้มีวิธีการประเมินที่ดีขึ้นแล้ว[ 117 ] [ 118 ]เมตริกที่อิงตามคำอธิบายประกอบเป็นการประเมินความสมบูรณ์ของการประกอบที่ดีกว่า เช่นจำนวนการจับคู่ที่ดีที่สุดของคอนทิก เมื่อประกอบแบบ de novo แล้วการประกอบสามารถใช้เป็นข้อมูลอ้างอิงสำหรับวิธีการจัดเรียงลำดับในภายหลังและการวิเคราะห์การแสดงออกของยีนเชิงปริมาณ
| ซอฟต์แวร์ | ปล่อยแล้ว | อัปเดตล่าสุด | ประสิทธิภาพการคำนวณ | จุดแข็งและจุดอ่อน |
|---|---|---|---|---|
| โอเอซิสกำมะหยี่[ 119 ] [ 120 ] | 2008 | 2011 | ประสิทธิภาพต่ำ ทำงานแบบเธรดเดียว ต้องการ RAM สูง | โปรแกรมประกอบคำสั่งแบบอ่านสั้นดั้งเดิม ปัจจุบันถูกแทนที่ด้วยโปรแกรมอื่นที่มีบทบาทสำคัญกว่าแล้ว |
| SOAPdenovo-trans [ 109 ] | 2011 | 2014 | ปานกลาง, รองรับมัลติเธรด, ต้องการ RAM ระดับปานกลาง | นี่คือตัวอย่างแรกๆ ของโปรแกรมประกอบลำดับดีเอ็นเอแบบอ่านสั้น ซึ่งได้รับการปรับปรุงเพื่อใช้ในการประกอบลำดับจีโนมแล้ว |
| ทรานส์-เอบีเอสเอส[ 121 ] | 2010 | 2016 | ปานกลาง, รองรับมัลติเธรด, ต้องการ RAM ระดับปานกลาง | เหมาะสำหรับลำดับเบสสั้น สามารถจัดการกับทรานสคริปโทมที่ซับซ้อนได้ และมีเวอร์ชันแบบขนาน MPI สำหรับคลัสเตอร์คอมพิวเตอร์ |
| ตรีเอกภาพ[ 122 ] [ 97 ] | 2011 | 2017 | ปานกลาง, รองรับมัลติเธรด, ต้องการ RAM ระดับปานกลาง | เหมาะสำหรับข้อมูลลำดับสั้น สามารถจัดการกับข้อมูลทรานสคริปโทมที่ซับซ้อนได้ แต่ใช้หน่วยความจำมาก |
| miraEST [ 123 ] | 1999 | 2016 | ปานกลาง, รองรับมัลติเธรด, ต้องการ RAM ระดับปานกลาง | สามารถประมวลผลลำดับซ้ำๆ ผสมผสานรูปแบบการจัดลำดับที่แตกต่างกัน และรองรับแพลตฟอร์มการจัดลำดับที่หลากหลาย |
| นิวเบลอร์[ 124 ] | 2004 | 2012 | ต่ำ, เธรดเดียว, ต้องการ RAM สูง | ออกแบบมาเป็นพิเศษเพื่อรองรับข้อผิดพลาดในการจัดลำดับโฮโมพอลิเมอร์ ซึ่งเป็นลักษณะเฉพาะของเครื่องจัดลำดับ Roche 454 |
| เวิร์กเบนช์จีโนมิกส์ CLC [ 125 ] | 2008 | 2014 | ประสิทธิภาพสูง รองรับมัลติเธรด และใช้ RAM ต่ำ | มีอินเทอร์เฟซผู้ใช้แบบกราฟิก สามารถผสานรวมเทคโนโลยีการจัดลำดับที่หลากหลาย ไม่มีคุณสมบัติเฉพาะสำหรับทรานสคริปโตม และต้องซื้อใบอนุญาตก่อนใช้งาน |
| SPAdes [ 126 ] | 2012 | 2017 | ประสิทธิภาพสูง รองรับมัลติเธรด และใช้ RAM ต่ำ | ใช้สำหรับการทดลองทางด้านทรานสคริปโตมิกส์ในเซลล์เดี่ยว |
| RSEM [ 127 ] | 2011 | 2017 | ประสิทธิภาพสูง รองรับมัลติเธรด และใช้ RAM ต่ำ | สามารถประเมินความถี่ของทรานสคริปต์ที่มีการตัดต่อแบบทางเลือกได้ ใช้งานง่าย |
| เชือกผูก[ 98 ] [ 128 ] | 2015 | 2019 | ประสิทธิภาพสูง รองรับมัลติเธรด และใช้ RAM ต่ำ | สามารถใช้การผสมผสานระหว่างวิธีการประกอบลำดับดีเอ็นเอแบบอ้างอิงและแบบสร้างใหม่เพื่อระบุลำดับดีเอ็นเอได้ |
คำอธิบายสัญลักษณ์: RAM – หน่วยความจำเข้าถึงแบบสุ่ม; MPI – อินเทอร์เฟซการส่งข้อความ; EST – แท็กแสดงลำดับ
การวัดปริมาณ
การหาปริมาณการจัดเรียงลำดับอาจทำได้ในระดับยีน เอ็กซอน หรือทรานสคริปต์[ 91 ] [ 87 ]ผลลัพธ์ทั่วไปประกอบด้วยตารางจำนวนการอ่านสำหรับแต่ละคุณลักษณะที่ป้อนให้กับซอฟต์แวร์ ตัวอย่างเช่น สำหรับยีนใน ไฟล์ รูปแบบคุณลักษณะทั่วไปจำนวนการอ่านของยีนและเอ็กซอนสามารถคำนวณได้ค่อนข้างง่ายโดยใช้ HTSeq เป็นต้น[ 130 ]การหาปริมาณในระดับทรานสคริปต์มีความซับซ้อนมากขึ้นและต้องใช้วิธีการทางความน่าจะเป็นเพื่อประมาณความอุดมสมบูรณ์ของไอโซฟอร์มทรานสคริปต์จากข้อมูลการอ่านสั้น ตัวอย่างเช่น การใช้ซอฟต์แวร์ cufflinks [ 114 ]การอ่านที่จัดเรียงได้ดีเท่ากันในหลายตำแหน่งจะต้องถูกระบุและลบออก จัดเรียงไปยังตำแหน่งที่เป็นไปได้ตำแหน่งใดตำแหน่งหนึ่ง หรือจัดเรียงไปยังตำแหน่งที่มีความน่าจะเป็นมากที่สุด
วิธีการหาปริมาณบางวิธีสามารถหลีกเลี่ยงความจำเป็นในการจัดตำแหน่งการอ่านให้ตรงกับลำดับอ้างอิงได้อย่างแม่นยำ วิธีการของซอฟต์แวร์ kallisto รวมการจัดตำแหน่งเสมือนและการหาปริมาณเข้าไว้ในขั้นตอนเดียว ซึ่งทำงานได้เร็วกว่าวิธีการร่วมสมัย เช่น วิธีที่ใช้โดยซอฟต์แวร์ tophat/cufflinks ถึง 2 เท่า โดยมีภาระการคำนวณน้อยกว่า[ 131 ]
การแสดงออกที่แตกต่างกัน
เมื่อมีจำนวนเชิงปริมาณของแต่ละทรานสคริปต์แล้วการแสดงออกของยีนที่แตกต่างกันจะถูกวัดโดยการทำให้เป็นมาตรฐาน สร้างแบบจำลอง และวิเคราะห์ข้อมูลทางสถิติ[ 108 ]เครื่องมือส่วนใหญ่จะอ่านตารางยีนและจำนวนการอ่านเป็นอินพุต แต่บางโปรแกรม เช่น cuffdiff จะยอมรับการจัด เรียงลำดับการอ่านในรูป แบบแผนที่การจัดเรียงแบบไบนารีเป็นอินพุต ผลลัพธ์สุดท้ายของการวิเคราะห์เหล่านี้คือรายการยีนพร้อมการทดสอบแบบจับคู่ที่เกี่ยวข้องสำหรับการแสดงออกที่แตกต่างกันระหว่างการรักษาและการประมาณความน่าจะเป็นของความแตกต่างเหล่านั้น[ 132 ]
| ซอฟต์แวร์ | สิ่งแวดล้อม | ความเชี่ยวชาญเฉพาะด้าน |
|---|---|---|
| Cuffdiff2 [ 108 ] | อิงตามระบบยูนิก | การวิเคราะห์การถอดรหัสที่ติดตามการตัดต่อทางเลือกของ mRNA |
| EdgeR [ 93 ] | อาร์/ไบโอคอนดักเตอร์ | ข้อมูลจีโนมที่นับจำนวนใดๆ |
| DEseq2 [ 133 ] | อาร์/ไบโอคอนดักเตอร์ | รูปแบบข้อมูลที่ยืดหยุ่น การทำสำเนาข้อมูลต่ำ |
| ลิมมา/วูม[ 92 ] | อาร์/ไบโอคอนดักเตอร์ | ข้อมูลไมโครอาร์เรย์หรือ RNA-Seq ช่วยให้สามารถออกแบบการทดลองได้อย่างยืดหยุ่น |
| ชุดราตรี[ 134 ] | อาร์/ไบโอคอนดักเตอร์ | การค้นหาข้อมูลถอดเสียงที่มีประสิทธิภาพและละเอียดอ่อน ยืดหยุ่นได้ |
คำอธิบายสัญลักษณ์: mRNA - อาร์เอ็นเอส่งสาร (messenger RNA)
การตรวจสอบความถูกต้อง
การวิเคราะห์ทรานสคริปโตมิกอาจได้รับการตรวจสอบความถูกต้องโดยใช้เทคนิคอิสระ เช่น qPCR ( quantitative PCR ) ซึ่งเป็นที่รู้จักและประเมินทางสถิติได้[ 135 ]การแสดงออกของยีนจะถูกวัดเทียบกับมาตรฐานที่กำหนดไว้สำหรับทั้งยีนที่สนใจและ ยีน ควบคุมการวัดโดย qPCR นั้นคล้ายกับที่ได้จาก RNA-Seq ซึ่งสามารถคำนวณค่าความเข้มข้นของบริเวณเป้าหมายในตัวอย่างที่กำหนดได้ อย่างไรก็ตาม qPCR ถูกจำกัดไว้ที่แอมพลิคอนที่มีขนาดเล็กกว่า 300 bp โดยปกติจะอยู่ทางด้าน 3' ของบริเวณการเข้ารหัส หลีกเลี่ยง3'UTR [ 136 ]หากจำเป็นต้องตรวจสอบความถูกต้องของไอโซฟอร์มของทรานสคริปต์ การตรวจสอบการจัดเรียงลำดับการอ่าน RNA-Seq ควรระบุตำแหน่งที่ อาจวาง ไพรเมอร์ qPCR เพื่อให้ ได้ความแตกต่างสูงสุด การวัดยีนควบคุมหลายตัวพร้อมกับยีนที่สนใจจะสร้างข้อมูลอ้างอิงที่เสถียรภายในบริบททางชีววิทยา[ 137 ]การตรวจสอบความถูกต้องของข้อมูล RNA-Seq ด้วย qPCR โดยทั่วไปแสดงให้เห็นว่าวิธีการ RNA-Seq ที่แตกต่างกันมีความสัมพันธ์กันสูง[ 62 ] [ 138 ] [ 139 ]
การตรวจสอบความถูกต้องเชิงฟังก์ชันของยีนสำคัญถือเป็นข้อพิจารณาที่สำคัญสำหรับการวางแผนหลังทรานสคริปโตม รูปแบบการแสดงออกของยีนที่สังเกตได้อาจเชื่อมโยงกับฟีโนไทป์โดย การศึกษา การลด / กู้คืนอิสระในสิ่งมีชีวิตที่สนใจ[ 140 ]
แอปพลิเคชัน
การวินิจฉัยและการวิเคราะห์ลักษณะเฉพาะของโรค
กลยุทธ์ทรานสคริปโตมิกส์ได้รับการ ประยุกต์ใช้อย่างกว้างขวางในหลากหลายสาขาของการวิจัยทางการแพทย์ชีวภาพ รวมถึงการวินิจฉัย โรค และการสร้างโปรไฟล์ [ 10 ] [ 141 ] แนวทาง RNA-Seq ช่วยให้สามารถระบุ ตำแหน่งเริ่มต้นการถอดรหัสในวงกว้างค้นพบ การใช้ โปรโมเตอร์ ทางเลือก และการเปลี่ยนแปลงการต่อเชื่อม แบบใหม่ องค์ประกอบควบคุมเหล่านี้มีความสำคัญในโรคของมนุษย์ ดังนั้น การกำหนดตัวแปรดังกล่าวจึงมีความสำคัญต่อการตีความการศึกษาที่เกี่ยวข้องกับโรค [ 142 ] RNA -Seq ยังสามารถระบุโพลีมอร์ฟิซึมของนิวคลีโอไทด์เดี่ยว (SNP) ที่เกี่ยวข้องกับโรค การแสดงออกเฉพาะอัลลีล และการหลอมรวมยีนซึ่งมีส่วนช่วยในการทำความเข้าใจตัวแปรที่ก่อให้เกิดโรค[ 143 ]
เรโทรทรานสโพซอนเป็นองค์ประกอบที่เคลื่อนย้ายได้ซึ่งแพร่กระจายภายในจีโนมของยูคาริโอตผ่านกระบวนการ ที่เกี่ยวข้องกับ การถอดรหัสย้อนกลับ RNA-Seq สามารถให้ข้อมูลเกี่ยวกับการถอดรหัสของเรโทรทรานสโพซอนภายในที่อาจมีอิทธิพลต่อการถอดรหัสของยีนข้างเคียงโดยกลไกเอพิเจเนติกส์ ต่างๆ ที่นำไปสู่โรค[ 144 ]ในทำนองเดียวกัน ศักยภาพในการใช้ RNA-Seq เพื่อทำความเข้าใจโรคที่เกี่ยวข้องกับภูมิคุ้มกันกำลังขยายตัวอย่างรวดเร็วเนื่องจากความสามารถในการวิเคราะห์ประชากรเซลล์ภูมิคุ้มกันและจัด ลำดับเรพเพอร์ทัวร์ของ เซลล์ Tและเซลล์ Bจากผู้ป่วย[ 145 ] [ 146 ]
ทรานสคริปโตมของมนุษย์และเชื้อโรค
RNA-Seq ของเชื้อก่อโรค ในมนุษย์ ได้กลายเป็นวิธีการที่ได้รับการยอมรับในการวัดปริมาณการเปลี่ยนแปลงการแสดงออกของยีน การระบุปัจจัยก่อโรค ใหม่ การทำนายความต้านทานยาปฏิชีวนะและการเปิดเผยปฏิสัมพันธ์ทางภูมิคุ้มกันระหว่างโฮสต์และเชื้อก่อโรค [ 147 ] [ 148 ] เป้าหมายหลักของเทคโนโลยีนี้คือการพัฒนา มาตรการ ควบคุมการติดเชื้อ ที่เหมาะสมที่สุด และการรักษาเฉพาะบุคคลที่ ตรงเป้าหมาย [ 146 ]
การวิเคราะห์ทรานสคริปโตมิกส่วนใหญ่มุ่งเน้นไปที่โฮสต์หรือเชื้อโรคเป็นหลัก เทคนิค Dual RNA-Seq ถูกนำมาใช้เพื่อสร้างโปรไฟล์การแสดงออกของ RNA ทั้งในเชื้อโรคและโฮสต์ตลอดกระบวนการติดเชื้อ เทคนิคนี้ช่วยให้สามารถศึกษาการตอบสนองแบบไดนามิกและเครือข่ายการควบคุมยีน ระหว่าง สายพันธุ์ในคู่ปฏิสัมพันธ์ทั้งสอง ตั้งแต่การสัมผัสครั้งแรกไปจนถึงการบุกรุกและการคงอยู่ของเชื้อโรคหรือการกำจัดโดยระบบภูมิคุ้มกันของ โฮสต์ในที่สุด [ 149 ] [ 150 ]
การตอบสนองต่อสิ่งแวดล้อม
ทรานสคริปโตมิกส์ช่วยให้สามารถระบุยีนและวิถีการทำงานที่ตอบสนองและต่อต้าน ความเครียดจากสิ่งแวดล้อมทั้ง ทางชีวภาพและทางกายภาพได้[ 151 ] [ 140 ]ลักษณะที่ไม่เจาะจงเป้าหมายของทรานสคริปโตมิกส์ช่วยให้สามารถระบุเครือข่ายการถอดรหัสทางพันธุกรรมแบบใหม่ในระบบที่ซับซ้อนได้ ตัวอย่างเช่น การวิเคราะห์เปรียบเทียบสายพันธุ์ถั่วชิกพี หลาย สายพันธุ์ในระยะการพัฒนาที่แตกต่างกันได้ระบุโปรไฟล์การถอดรหัสทางพันธุกรรมที่แตกต่างกันซึ่งเกี่ยวข้องกับ ความเครียด จากภัยแล้งและความเค็มรวมถึงการระบุบทบาทของไอโซฟอร์มการถอดรหัสของAP2 - EREBP [ 151 ]การตรวจสอบการแสดงออกของยีนในระหว่าง การก่อตัว ของไบโอฟิล์มโดยเชื้อราก่อโรคCandida albicansเผยให้เห็นชุดยีนที่ควบคุมร่วมกันซึ่งมีความสำคัญต่อการสร้างและการบำรุงรักษาไบโอฟิล์ม[ 152 ]
การวิเคราะห์โปรไฟล์ทรานสคริปโตมิกยังให้ข้อมูลที่สำคัญเกี่ยวกับกลไกการดื้อยาอีกด้วย การวิเคราะห์เชื้อPlasmodium falciparum มากกว่า 1,000 ตัวอย่าง ซึ่งเป็นปรสิตที่ก่อให้เกิดโรคมาลาเรียในมนุษย์[ 153 ]พบว่าการเพิ่มขึ้นของการตอบสนองของโปรตีนที่พับตัวผิดปกติ และความก้าวหน้าที่ช้าลงในช่วงเริ่มต้นของ วงจรการพัฒนาภายในเม็ดเลือดแดงแบบไม่อาศัยเพศนั้นเกี่ยวข้องกับความต้านทานต่ออาร์เทมิซินินในเชื้อที่แยกได้จากเอเชียตะวันออกเฉียงใต้ [ 154 ]
การใช้ทรานสคริปโตมิกส์มีความสำคัญต่อการศึกษาการตอบสนองในสภาพแวดล้อมทางทะเล เช่นกัน [ 155 ]ในนิเวศวิทยาทางทะเล " ความเครียด " และ " การปรับตัว " เป็นหัวข้อวิจัยที่พบได้บ่อยที่สุด โดยเฉพาะอย่างยิ่งที่เกี่ยวข้องกับความเครียดจากกิจกรรมของมนุษย์ เช่นการเปลี่ยนแปลงสภาพภูมิอากาศโลกและมลภาวะ [ 155 ]การศึกษาส่วนใหญ่ในด้านนี้ทำในสัตว์แม้ว่าสัตว์ไม่มีกระดูกสันหลังจะได้รับ การศึกษาน้อย [ 155 ]ปัญหาหนึ่งที่ยังคงมีอยู่คือ การขาดแคลนการศึกษาทางพันธุศาสตร์เชิงหน้าที่ ซึ่งขัดขวาง การ ระบุยีนโดยเฉพาะอย่างยิ่งสำหรับสายพันธุ์ที่ไม่ใช่แบบจำลอง และอาจนำไปสู่ข้อสรุปที่ไม่ชัดเจนเกี่ยวกับผลกระทบของการตอบสนองที่ศึกษา[ 155 ]
ทรานสคริปโตมิกส์มีประโยชน์อย่างยิ่งในการศึกษาว่ายีนของแบคทีเรียตอบสนองต่อสภาพแวดล้อมที่ซับซ้อนซึ่งเกี่ยวข้องกับพืชอย่างไร โดยที่การปรับตัวนั้นเกิดจากการควบคุมที่ประสานกันของยีนหลายตัวมากกว่าปัจจัยเดียวที่ได้รับการระบุอย่างชัดเจน ในปฏิสัมพันธ์ระหว่างพืชและจุลินทรีย์ การวิเคราะห์ทรานสคริปโตมิกส์ได้ระบุชุดของยีนแบคทีเรียที่ถูกกระตุ้นโดยเฉพาะในระหว่างการเข้าอาศัยของพืชหรือในการตอบสนองต่อสัญญาณที่มาจากพืช ซึ่งหลายยีนนั้นขาดการระบุ หน้าที่มาก่อน และจะไม่สามารถตรวจพบได้โดยใช้แนวทางที่มุ่งเป้าหมายเฉพาะ การบูรณาการข้อมูลการแสดงออกของยีนกับ การวิเคราะห์ จีโนมเปรียบเทียบหรือการวิเคราะห์ระดับชุมชน ทรานสคริปโตมิกส์ช่วยเชื่อมโยงการควบคุมยีนของแบคทีเรียเข้ากับความเกี่ยวข้องทางนิเวศวิทยาและกระบวนการปรับตัวในแหล่งที่อยู่อาศัยที่เกี่ยวข้องกับพืช แนวทางนี้พิสูจน์แล้วว่ามีคุณค่าในการระบุหน้าที่ของแบคทีเรียที่ถูกมองข้ามไปก่อนหน้านี้ ซึ่งเกี่ยวข้องกับการทนต่อความเครียด ปฏิสัมพันธ์กับโฮสต์ และการปรับตัวเข้ากับแหล่งที่อยู่อาศัย
การระบุหน้าที่ของยีน
เทคนิคทรานสคริปโตมิกทั้งหมดมีประโยชน์อย่างยิ่งในการระบุหน้าที่ของยีนและระบุยีนที่รับผิดชอบต่อฟีโนไทป์เฉพาะ ทรานสคริปโตมิกของสายพันธุ์Arabidopsis ที่สะสมโลหะมากเกินไปมีความสัมพันธ์ระหว่างยีนที่เกี่ยวข้องกับการดูดซึมโลหะความทนทาน และภาวะสมดุลกับฟีโนไทป์[ 156 ]การบูรณาการชุดข้อมูล RNA-Seq จากเนื้อเยื่อต่างๆ ได้ถูกนำมาใช้เพื่อปรับปรุงการระบุหน้าที่ของยีนในสิ่งมีชีวิตที่มีความสำคัญทางการค้า (เช่นแตงกวา ) [ 157 ]หรือสายพันธุ์ที่ใกล้สูญพันธุ์ (เช่นโคอาลา ) [ 158 ]
การประกอบลำดับ RNA-Seq ไม่ขึ้นอยู่กับจีโนมอ้างอิง[ 122 ]ดังนั้นจึงเหมาะสำหรับการศึกษาการแสดงออกของยีนในสิ่งมีชีวิตที่ไม่ใช่แบบจำลองที่มีทรัพยากรจีโนมที่ไม่มีอยู่หรือพัฒนาได้ไม่ดี ตัวอย่างเช่น ฐานข้อมูล SNP ที่ใช้ในโปรแกรม การผสมพันธุ์ ต้นสนดักลาสถูกสร้างขึ้นโดย การวิเคราะห์ทรานสคริปโตมแบบ de novoในกรณีที่ไม่มีจีโนมที่ได้รับการจัดลำดับ[ 159 ]ในทำนองเดียวกัน ยีนที่ทำหน้าที่ในการพัฒนาเนื้อเยื่อหัวใจ กล้ามเนื้อ และระบบประสาทในกุ้งมังกรถูกระบุโดยการเปรียบเทียบทรานสคริปโตมของเนื้อเยื่อประเภทต่างๆ โดยไม่ต้องใช้ลำดับจีโนม[ 160 ] RNA-Seq ยังสามารถใช้เพื่อระบุบริเวณการเข้ารหัสโปรตีน ที่ไม่เคยรู้จักมาก่อน ในจีโนมที่ได้รับการจัดลำดับที่มีอยู่
อาร์เอ็นเอที่ไม่เข้ารหัส
ทรานสคริปโตมิกส์มักถูกนำไปใช้กับเนื้อหา mRNA ของเซลล์เป็นส่วนใหญ่ อย่างไรก็ตาม เทคนิคเดียวกันนี้ก็สามารถนำไปใช้กับ RNA ที่ไม่เข้ารหัส (ncRNA) ได้เช่นกัน ซึ่ง RNA เหล่านั้นไม่ได้ถูกแปลเป็นโปรตีน แต่มีหน้าที่โดยตรง (เช่น บทบาทในการแปลโปรตีนการจำลองDNA การตัดต่อ RNAและการควบคุมการถอดรหัส ) [ 161 ] [ 162 ] [ 163 ] [ 164 ] ncRNA เหล่านี้จำนวนมากส่งผลต่อสภาวะของโรคต่างๆ รวมถึงมะเร็ง โรคหัวใจและหลอดเลือด และโรคทางระบบประสาท[ 165 ]
ฐานข้อมูลทรานสคริปโตม
การศึกษาทรานสคริปโตมิกส์สร้างข้อมูลจำนวนมากที่มีศักยภาพในการประยุกต์ใช้ที่กว้างไกลเกินกว่าเป้าหมายดั้งเดิมของการทดลอง ดังนั้น ข้อมูลดิบหรือข้อมูลที่ประมวลผลแล้วอาจถูกฝากไว้ในฐานข้อมูลสาธารณะเพื่อให้มั่นใจได้ว่าข้อมูลเหล่านั้นจะมีประโยชน์สำหรับชุมชนวิทยาศาสตร์ในวงกว้าง ตัวอย่างเช่น ในปี 2018 Gene Expression Omnibus มีการทดลองนับล้านรายการ[ 166 ]
| ชื่อ | เจ้าภาพ | ข้อมูล | คำอธิบาย |
|---|---|---|---|
| รถโดยสารการแสดงออกของยีน[ 100 ] | เอ็นซีบีไอ | ไมโครอาร์เรย์ RNA-Seq | ฐานข้อมูลทรานสคริปโตมิกส์แรกที่ยอมรับข้อมูลจากแหล่งใดก็ได้ นำเสนอ มาตรฐานชุมชน MIAMEและMINSEQEที่กำหนดเมตาเดตาการทดลองที่จำเป็นเพื่อให้มั่นใจถึงการตีความที่มีประสิทธิภาพและความสามารถในการทำซ้ำ[ 167 ] [ 168 ] |
| ArrayExpress [ 169 ] | อีนา | ไมโครอาร์เรย์ | นำเข้าชุดข้อมูลจาก Gene Expression Omnibus และรับการส่งข้อมูลโดยตรง ข้อมูลที่ประมวลผลแล้วและข้อมูลเมตาของการทดลองจะถูกจัดเก็บไว้ที่ ArrayExpress ในขณะที่ลำดับการอ่านดิบจะถูกเก็บไว้ที่ ENA เป็นไปตามมาตรฐาน MIAME และ MINSEQE [ 167 ] [ 168 ] |
| แอตลาสการแสดงออก[ 170 ] | อีบีไอ | ไมโครอาร์เรย์ RNA-Seq | ฐานข้อมูลการแสดงออกของยีนเฉพาะเนื้อเยื่อสำหรับสัตว์และพืช แสดงผลการวิเคราะห์ขั้นที่สองและการแสดงภาพ เช่น การเสริมคุณค่าเชิงหน้าที่ของ คำศัพท์ Gene Ontology , โดเมน InterProหรือวิถีการทำงานต่างๆ เชื่อมโยงไปยังข้อมูลปริมาณโปรตีนหากมี |
| Genevestigator [ 171 ] | คัดสรรโดยเอกชน | ไมโครอาร์เรย์ RNA-Seq | ประกอบด้วยการคัดกรองข้อมูลทรานสคริปโตมสาธารณะด้วยตนเอง โดยเน้นข้อมูลด้านชีววิทยาทางการแพทย์และพืช การทดลองแต่ละครั้งจะถูกปรับให้เป็นมาตรฐานทั่วทั้งฐานข้อมูล เพื่อให้สามารถเปรียบเทียบการแสดงออกของยีนในการทดลองที่หลากหลายได้ การใช้งานฟังก์ชันเต็มรูปแบบต้องซื้อใบอนุญาต โดยสามารถเข้าถึงฟังก์ชันที่จำกัดได้ฟรี |
| RefEx [ 172 ] | ดีดีบีเจ | ทั้งหมด | ข้อมูลการถอดรหัสพันธุกรรมของมนุษย์ หนู และหนูแรต จาก 40 อวัยวะที่แตกต่างกัน การแสดงออกของยีนแสดงผลในรูปแบบแผนที่ความร้อนที่ฉายลงบนแบบจำลอง 3 มิติของโครงสร้างทางกายวิภาค |
| นอกรหัส[ 173 ] | นอนโค้ด.org | RNA-Seq | อาร์เอ็นเอที่ไม่เข้ารหัส (ncRNA) ซึ่งไม่รวมถึง tRNA และ rRNA |
คำอธิบายสัญลักษณ์: NCBI – ศูนย์ข้อมูลเทคโนโลยีชีวภาพแห่งชาติ; EBI – สถาบันชีวสารสนเทศแห่งยุโรป; DDBJ – ธนาคารข้อมูลดีเอ็นเอแห่งประเทศญี่ปุ่น; ENA – คลังข้อมูลนิวคลีโอไทด์แห่งยุโรป; MIAME – ข้อมูลขั้นต่ำเกี่ยวกับการทดลองไมโครอาร์เรย์; MINSEQE – ข้อมูลขั้นต่ำเกี่ยวกับการทดลองการจัดลำดับนิวคลีโอไทด์ความเร็วสูง
ดูเพิ่มเติม
อ่านเพิ่มเติม
- Lowe R, Shirley N, Bleackley M, Dolan S, Shafee T (พฤษภาคม 2017). "เทคโนโลยีทรานสคริปโตมิกส์" . PLOS Computational Biology . 13 (5) e1005457. Bibcode : 2017PLSCB..13E5457L . doi : 10.1371/journal.pcbi.1005457 . PMC 5436640 . PMID 28545146 .
- การวิเคราะห์ทรานสคริปโตมิกส์เชิงเปรียบเทียบในโมดูลอ้างอิงในวิทยาศาสตร์ชีวภาพ
- ซอฟต์แวร์ที่ใช้ในงานทรานสคริปโตมิกส์:
- กระดุมข้อมือ
- คัลลิสโต
- หมวกทรงสูง