อ่าน 22 นาที
ลิโปซาร์โคมา
ลิโปซาร์โคมาเป็นชนิดย่อยที่พบบ่อยที่สุดของซาร์โคมา เนื้อเยื่ออ่อน คิดเป็นอย่างน้อย 20% ของซาร์โคมาทั้งหมดในผู้ใหญ่ซาร์โคมาเนื้อเยื่ออ่อนเป็นเนื้องอก ที่หายาก มี...
ลิโปซาร์โคมา
| ลิโปซาร์โคมา | |
|---|---|
 | |
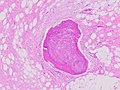
| พยาธิวิทยาของลิโปซาร์โคมา การย้อมสี H&E: [ 1 ] - | |
| ความเชี่ยวชาญ | โรคผิวหนังศัลยกรรมทั่วไป และ มะเร็งวิทยา |
| อาการ | ก้อนใต้ผิวหนัง ปวด บวม การทำงานของอวัยวะผิดปกติ |
ลิโปซาร์โคมาเป็นชนิดย่อยที่พบบ่อยที่สุดของซาร์โคมา เนื้อเยื่ออ่อน คิดเป็นอย่างน้อย 20% ของซาร์โคมาทั้งหมดในผู้ใหญ่[ 2 ]ซาร์โคมาเนื้อเยื่ออ่อนเป็นเนื้องอก ที่หายาก มี ชนิดย่อยหรือรูปแบบทางเนื้อเยื่อวิทยามากกว่า 150 ชนิด ลิโปซาร์โคมาเกิดขึ้นจาก ลิโปบลาสต์ซึ่งเป็นเซลล์ไขมันในเนื้อเยื่อไขมันเนื้อเยื่อไขมันกระจายอยู่ทั่วร่างกาย รวมถึงบริเวณต่างๆ เช่น ชั้นใต้ผิวหนัง ที่ลึกและตื้นกว่า ตลอดจนบริเวณที่เข้าถึงได้ยากในการผ่าตัด เช่นช่องว่างหลังช่องท้อง ( retroperitoneum ) และไขมันในช่องท้อง[ 3 ]
ลิโปซาร์โคมาทั้งหมดประกอบด้วยเซลล์อย่างน้อยบางส่วนที่มีลักษณะคล้ายเซลล์ไขมันเมื่อตรวจสอบ ลักษณะ ทางจุลพยาธิวิทยาภายใต้กล้องจุลทรรศน์[ 4 ]อย่างไรก็ตาม ลิโปซาร์โคมามีหลายรูปแบบขึ้นอยู่กับความแตกต่างในการนำเสนอทางคลินิก (เช่น อายุ เพศ ตำแหน่งของเนื้องอกอาการและสัญญาณ ) ความรุนแรง (เช่น ศักยภาพในการรุกรานเนื้อเยื่อในบริเวณใกล้เคียง การกลับมาเป็นซ้ำหลังการผ่าตัด และการแพร่กระจายไปยังเนื้อเยื่อส่วนปลาย) ความผิดปกติทางพันธุกรรม การพยากรณ์โรคและวิธีการรักษาที่ต้องการองค์การอนามัยโลกในปี 2020 ได้จัดประเภทลิโปซาร์โคมาใหม่เป็น 5 รูปแบบที่แตกต่างกันไม่มากก็น้อย ได้แก่1)เนื้องอกไขมันผิดปกติ/ลิโปซาร์โคมาที่มีการแยกแยะเซลล์ได้ดี (WD-LPS); 2)ลิโปซาร์โคมาที่มีการแยกแยะเซลล์ลดลง (DD-LPS); 3)ลิโปซาร์โคมาชนิดเมือก; 4)ลิโปซาร์โคมาชนิดหลายรูปแบบ; และ5)ไลโปซาร์โคมาชนิดไมซอยด์พลีโอโมฟิก[ 5 ] ( พลีโอโมฟิกหมายถึงการมีเซลล์ที่มีขนาดและรูปร่างผิดปกติและมักมีความแตกต่างกันมาก และ/หรือขนาดและรูปร่างของนิวเคลียส)
แม้ว่าลิโปซาร์โคมาจะถูกจัดประเภทเป็นเนื้องอกที่ก้าวร้าวและร้ายแรงหรือในกรณีของเนื้องอกไขมันผิดปกติ/ลิโปซาร์โคมาที่มีการแยกแยะเซลล์ได้ดี จะถูกจัดประเภทเป็นเนื้องอกที่ไม่ก้าวร้าวและไม่ร้ายแรง[ 6 ]แต่ลิโปซาร์โคมาทั้งห้าชนิดสามารถแทรกซึมเข้าไปในบริเวณใกล้เคียงเพื่อทำลายเนื้อเยื่อและอวัยวะที่อยู่ใกล้เคียง เกิดขึ้นในบริเวณที่เข้าถึงได้ยากทางศัลยกรรมที่อยู่ติดกับอวัยวะสำคัญ (เช่น ช่องท้องส่วนหลัง[ 7 ] ) เกิดการกำเริบซ้ำหลังจากการผ่าตัดเอาออก และลุกลามไปสู่โรคที่คุกคามชีวิต การศึกษาจนถึงปัจจุบันพบว่าลิโปซาร์โคมาทั้งห้าชนิด แม้ว่าจะสามารถรักษาได้อย่างน้อยในเบื้องต้นด้วยการผ่าตัดเอาออก แต่ก็มักจะตอบสนองต่อเคมีบำบัดและรังสีบำบัด ที่ใช้ในปัจจุบันเพียงเล็กน้อยเท่านั้น ลิโปซาร์โคมาจำเป็นต้องมีการศึกษาเพิ่มเติมอีกมากมายเพื่อพิจารณาการตอบสนองต่อรังสีบำบัดเคมีบำบัดและวิธีการรักษาใหม่ๆ ที่ใช้แยกกันและใช้ร่วมกันในรูปแบบต่างๆ ซึ่งจะรวมถึงการผ่าตัดเอาออกหากเป็นไปได้[ 6 ]
นิรุกติศาสตร์
"เนื้องอกไขมัน" (พหูพจน์ lipomata) ค.ศ. 1830 มาจากภาษาละตินทางการแพทย์ มาจากภาษากรีก lipos "ไขมัน" (คำนาม) มาจากรากศัพท์ PIE *leip- "ติด, เกาะ" ซึ่งใช้ในการสร้างคำที่หมายถึง "ไขมัน" เช่นกัน + -oma
ทศวรรษ 1650 "ติ่งเนื้อ" (พหูพจน์ liposarcomata) มาจากภาษาละตินทางการแพทย์ ซึ่งมาจากรูปภาษาละตินของคำภาษากรีก sarkoma "สารเนื้อ" (Galen) มาจาก sarkoun "สร้างเนื้อ, เติบโตเป็นเนื้อ" มาจาก sarx (รูปกรรมวาจก sarkos) "เนื้อ" + -oma
รูปแบบของลิโปซาร์โคมา

โดยทั่วไปแล้วลิโปซาร์โคมาเป็นเนื้องอกขนาดใหญ่ (>10 ซม.) แต่สามารถมีขนาดได้เกือบทุกขนาด ส่วนใหญ่เกิดขึ้นในผู้ใหญ่ โดยมีเพียง 0.7% ของกรณีที่เกิดขึ้นในกลุ่มอายุต่ำกว่า 16 ปี[ 5 ]ในผู้ใหญ่ ลิโปซาร์โคมาส่วนใหญ่มักเกิดขึ้นในวัยกลางคนขึ้นไป[ 8 ]กรณีที่พบได้น้อยมากในเด็กและวัยรุ่น มักได้รับการวินิจฉัยว่าเป็นลิโปซาร์โคมาชนิดมิกซอยด์[ 5 ]
ลิโปซาร์โคมาทั้งห้าชนิดจะต้องแยกแยะออกจากกันเองและจากเนื้องอกเนื้อเยื่ออ่อนชนิดอื่นๆ ด้วย เนื้องอกชนิดอื่นๆ เหล่านี้พร้อมกับลักษณะทางพยาธิวิทยาที่แตกต่างกันบางประการ ได้แก่: 1)ลิโปมาดิสพลาสติก (เช่น เนื้องอกที่ไม่ร้ายแรงที่มีบริเวณเนื้อเยื่อตาย และเซลล์ไขมันที่เป็นเนื้องอกขนาดต่างๆ ที่มี นิวเคลียสขนาด/รูปร่างต่างๆ กันเซลล์เนื้องอกเหล่านี้ ซึ่งแตกต่างจากเซลล์เนื้องอกส่วนใหญ่ในลิโปซาร์โคมา ไม่แสดงออก มากเกินไป ของ ยีน MDM2 ); [ 9 ] 2) ลิโปมาเซลล์รูปทรงกระสวยที่ผิดปกติ(เช่น เนื้องอกที่ไม่ร้ายแรงที่มีเซลล์รูปทรงกระสวยที่ผิดปกติเล็กน้อยในสโตรมา ที่มีลักษณะเป็นเส้นใยถึงเมือก ผสมกับลิโปบลาสต์ ที่มีช่องว่าง และเซลล์ไขมันขนาดต่างๆ ที่มีนิวเคลียสที่ผิดปกติ; 3) ลิโปมาพลีโอโมฟิก (เช่น เนื้องอกที่ไม่ร้ายแรงที่มีลักษณะเป็นเซลล์ขนาดใหญ่ที่มีนิวเคลียสซ้อนทับกัน); [ 8 ]และ4) เนื้องอกเส้นใยเดี่ยว (เช่น เนื้องอกที่มีพฤติกรรมร้ายแรงมากถึง 22% ประกอบด้วยเซลล์รูปทรงกระสวยหรือรูปไข่ภายในเนื้อเยื่อเกี่ยวพันที่เป็นคอลลาเจนผสมกับหลอดเลือดที่มีรูปร่างคล้ายเขากวาง[ 10 ] ) [ 5 ]
เนื้องอกไขมันชนิดผิดปกติ/มะเร็งไขมันชนิดที่มีการแยกแยะเซลล์ได้ดี
โดยรวมแล้ว เนื้องอกไขมันผิดปกติ (ALTs) และลิโปซาร์โคมาที่มีการแยกแยะเซลล์ได้ดี (WDLs) คิดเป็น 40–45% ของลิโปซาร์โคมาทั้งหมด[ 11 ] เนื้องอก เหล่านี้แทบจะไม่แพร่กระจายไป ยังส่วนอื่นของร่างกาย เลย ดังนั้นจึงถือว่าเป็นเนื้องอกที่ไม่ร้ายแรงหรือเนื้องอกก่อนเป็นมะเร็ง[ 12 ] [ 13 ]อย่างไรก็ตาม เนื้องอกเหล่านี้มีการรุกรานเฉพาะที่และอาจกลายพันธุ์เป็นลิโปซาร์โคมาที่รุนแรงกว่าและอาจแพร่กระจายไปยังส่วนอื่นของร่างกายได้ เช่น ลิโปซาร์โคมาที่มีการแยกแยะเซลล์ได้แย่ลง นอกจากนี้ เนื้องอกไขมันผิดปกติ/ลิโปซาร์โคมาที่มีการแยกแยะเซลล์ได้ดีที่ถูกผ่าตัดออกไปแล้ว อาจกลับมาเป็นลิโปซาร์โคมาที่มีการแยกแยะเซลล์ได้แย่ลงอีกครั้ง[ 6 ]
การนำเสนอ

ALT และ WDL ถือเป็นเนื้องอกที่แทบจะเหมือนกันทุกประการ ยกเว้นว่าตามคำจำกัดความ ALT หมายถึงเนื้องอกที่เกิดขึ้นในแขนหรือขา ในขณะที่ WDL หมายถึงเนื้องอกที่เกิดขึ้นในบริเวณที่เข้าถึงได้ยากกว่าในการผ่าตัด เช่น เนื้อเยื่ออ่อนที่อยู่ลึกและอยู่ตรงกลางของ ช่องท้องส่วนหลัง บริเวณ รอบอัณฑะ (เช่น บริเวณภายในถุงอัณฑะรวมถึงอัณฑะสายอสุจิเยื่อหุ้มอัณฑะ เอ พิเดิดิมิสและ ส่วนต่อ ท้ายของอัณฑะ ) [ 6 ]ช่องปากและเบ้าตา[ 12 ] [ 14 ]คำศัพท์นี้มีผลต่อการพยากรณ์โรค: น้อยกว่า 7% ของเนื้องอก ALT จะเปลี่ยนเป็นลิโปซาร์โคมาที่เสื่อมสภาพภายในระยะเวลาเฉลี่ย 7 ปี ในขณะที่ 17% ของเนื้องอก WDL จะเปลี่ยนเป็นลิโปซาร์โคมาที่เป็นมะเร็งร้ายแรงกว่านี้ภายในระยะเวลาเฉลี่ย 8 ปี[ 6 ]เนื้องอก ALT และ WDL (ต่อไปนี้เรียกว่า ALT/WDL) มักพบในผู้ที่มีอายุกลางคนและผู้สูงอายุ โดยมีลักษณะเป็นก้อนที่ค่อยๆ ขยายใหญ่ขึ้น และมีแนวโน้มที่จะมีขนาดใหญ่ขึ้นและอยู่ในระยะที่ลุกลามมากขึ้นเมื่ออยู่ในเนื้อเยื่อชั้นลึก[ 8 ] [ 11 ]เนื้องอกเหล่านี้มักไม่เจ็บปวด และหากอยู่ตื้นๆ ก็สามารถมองเห็นได้ง่าย นอกจากนี้ยังอาจทำให้เกิดอาการบวมน้ำ อย่างกว้างขวาง (เช่น อาการบวมเนื่องจากการสะสมของของเหลวในบริเวณนั้น) ในบริเวณที่เกี่ยวข้อง เช่น ต้นขา (ดูรูปที่อยู่ติดกัน) เนื่องจากการรุกรานเข้าไปในหลอดเลือดและ/หรือหลอดน้ำเหลืองที่ระบายจากบริเวณเนื้องอก เนื้องอก ALT/WDL ที่อยู่ลึกอาจไม่มีอาการ แต่ขึ้นอยู่กับตำแหน่งของมัน อาจทำให้เกิดสัญญาณและ/หรืออาการผิดปกติที่ร้ายแรงในอวัยวะต่างๆ ที่มันแทรกซึมเข้าไป อวัยวะเหล่านี้ได้แก่ อวัยวะที่อยู่ใกล้หรืออยู่ในช่องหลังเยื่อบุช่องท้อง (เช่น ลำไส้ ไต และท่อไต)และบริเวณรอบอัณฑะ ช่องอกส่วนกลาง (เช่นหลอดลมและหลอดลมใหญ่ ของปอด ) และศีรษะ (เช่น ช่องว่างหลังลูกตา) [ 12 ] [ 14 ]
พยาธิวิทยา

ในทางพยาธิวิทยาเนื้อเยื่อ เนื้องอก ALT/WDL แบ่งออกเป็นชนิดคล้ายเซลล์ไขมัน/ไลโปมา ชนิดแข็งตัว และชนิดอักเสบ โดยชนิดคล้ายเซลล์ไขมัน/ไลโปมาเป็นชนิดที่พบได้บ่อยที่สุด เนื้องอก ALT/WDL ชนิดคล้ายเซลล์ไขมัน/ไลโปมาประกอบด้วยกลุ่มเซลล์ไขมันที่เจริญเต็มที่ซึ่งแทรกด้วยเยื่อ เส้นใยที่ไม่สม่ำเสมอ (ดูภาพถ่ายจุลทรรศน์ ที่ ย้อมด้วย H&E ที่อยู่ด้านข้าง ) เนื้องอก ALT/WDL ชนิดแข็งตัว ซึ่งเป็นชนิดที่พบได้บ่อยเป็นอันดับสอง มักเกิดขึ้นในบริเวณหลังช่องท้องและรอบอัณฑะ ประกอบด้วยเซลล์เนื้อเยื่อเกี่ยวพัน ที่ผิดปกติกระจัดกระจายอยู่ภายใน เนื้อเยื่อเกี่ยวพัน ที่มี คอลลาเจน (เช่น มี คอลลาเจนเป็นส่วนประกอบ) เซลล์ไขมันอ่อน ที่มีช่องว่าง ภายในพบได้ น้อย ในเนื้อเยื่อนี้ เนื้องอก ALT/WDL ชนิดอักเสบเป็นชนิดที่พบได้ยากที่สุด มักพบได้บ่อยที่สุดในช่องท้องส่วนหลังและประกอบด้วย เซลล์ อักเสบเรื้อรังเช่นลิมโฟไซต์และเซลล์พลาสมารวมทั้งฟอลลิเคิลคล้ายต่อมน้ำเหลืองที่กระจายอยู่ทั่วพื้นหลังของเนื้อเยื่อที่มีเซลล์ไขมัน[ 14 ]
พันธุศาสตร์
เซลล์มะเร็งในเนื้องอก ALT/WDL มีโครโมโซมเครื่องหมายส่วนเกินรูปวงแหวนขนาดเล็ก (sSMC) หนึ่งตัวหรือมากกว่า หรือโครโมโซมเครื่องหมายขนาดใหญ่ที่ผิดปกติ (เช่น โครโมโซมปกติเดิมที่กลายเป็นผิดปกติโดยการทำซ้ำส่วนต่างๆ ของสารพันธุกรรมของตัวเองหรือของโครโมโซมอื่นหนึ่งตัวหรือมากกว่า) โครโมโซมที่ผิดปกติเหล่านี้มีสำเนาเพิ่มเติมของแขนยาวของโครโมโซม 12 (เรียกอีกอย่างว่า แขน q ) ที่แถบ 13 ถึง 15 ส่วนของโครโมโซม 12 นี้รวมถึงโปรโตออนโคยีนMDM2 (ยีนที่อาจก่อให้เกิดเนื้องอกเมื่อมีการแสดงออกมากเกินไป ) ซึ่งตั้งอยู่ที่แถบ 15 [ 15 ]และCDK4 (ยีนที่เมื่อมีการแสดงออกมากเกินไปจะส่งเสริมการพัฒนาของเนื้องอกต่างๆ) ซึ่งตั้งอยู่ที่แถบ 14.1 [ 16 ] [ 17 ]การขยาย (เช่น การเพิ่มจำนวนสำเนาของยีนโดยไม่มีการเพิ่มสัดส่วนของยีนอื่น) ของยีนทั้งสองนี้เป็นตัวบ่งชี้ที่มีความไวและความจำเพาะสูงว่าลิโปซาร์โคมาเป็น ALT/WDL หรือลิโปซาร์โคมาที่เสื่อมสภาพมากกว่าลิโปซาร์โคมาหรือลิโปมาชนิด อื่น [ 17 ]นอกจาก ยีน MDM2และCDK4แล้ว บริเวณโครโมโซมแถบ 13–15 นี้ยังประกอบด้วย ยีน TSPAN31และHMGA2ซึ่งเมื่อมีการแสดงออกมากเกินไป จะเกี่ยวข้องกับเนื้องอกและ/หรือมะเร็งต่างๆ มีการเสนอแนะว่ายีนที่มีการแสดงออกมากเกินไปอย่างน้อยหนึ่งยีนนี้ส่งเสริมและ/หรือมีส่วนช่วยในการพัฒนาและ/หรือความก้าวหน้าของเนื้องอก ALT/WDL [ 8 ]
การวินิจฉัย
การวินิจฉัยเนื้องอก ALT/WDL ทำขึ้นโดยพิจารณาจากลักษณะอาการทางคลินิก พยาธิวิทยา และผลการตรวจทางพันธุกรรม โดยเฉพาะอย่างยิ่ง การตรวจพบ ยีน MDM2หรือCDK4 ที่แสดงออกมากเกินไปในเซลล์เนื้องอก ALT/WDL หรือการมีอยู่ของโครโมโซมเครื่องหมาย sSMC หรือโครโมโซมเครื่องหมายยักษ์ที่เกี่ยวข้องกับ ALT/WDL โดยเฉพาะ (ตามที่กำหนดโดยการลำดับดีเอ็นเอรุ่นใหม่การ ผสม แบบเปรียบเทียบจีโน ม [ 18 ]และ/หรือการวิเคราะห์แถบ G ทางไซโตเจเนติกส์ที่มีความเชี่ยวชาญสูง[ 19 ] ) สนับสนุนการวินิจฉัย ALT/WDL หรือลิโปซาร์โคมาที่เสื่อมสภาพอย่างมาก ความแตกต่างของอาการทางคลินิกและพยาธิวิทยาของลิโปซาร์โคมาสองรูปแบบหลังนี้มักจะช่วยในการแยกแยะความแตกต่างระหว่างกันได้[ 8 ]
การรักษาและการพยากรณ์โรค
เนื้องอก ALT/WDL ได้รับการรักษาด้วยการผ่าตัดเอาเนื้อเยื่อเนื้องอกออกทั้งหมด อย่างไรก็ตาม เนื้องอกเหล่านี้จะกลับมาเป็นซ้ำในบริเวณเดิมใน 30–50% ของกรณี การกลับมาเป็นซ้ำมักเกิดขึ้นในเนื้องอกที่อยู่ในตำแหน่งที่เข้าถึงได้ยาก เช่น ในช่องท้องส่วนหลัง ช่องอก และท่ออสุจิ เนื้องอกที่เข้าถึงได้ยากทางการผ่าตัดเหล่านี้มีแนวโน้มที่จะกลับมาเป็นซ้ำหลายครั้งและในที่สุดอาจทำให้เสียชีวิตเนื่องจากมีผลเสียต่ออวัยวะสำคัญ แม้ว่าเนื้องอก ALT/WDL จะมีศักยภาพในการแพร่กระจาย น้อยมาก แต่ประมาณ 10% จะเปลี่ยนไปเป็นมะเร็งไขมันชนิดร้ายแรงและอาจแพร่กระจายได้ ซึ่งเรียกว่ามะเร็งไขมันชนิดที่เปลี่ยนแปลงสภาพ (dedifferentiated liposarcoma) ระยะเวลาเฉลี่ยสำหรับการเปลี่ยนแปลงเป็นมะเร็ง นี้ อยู่ที่ประมาณ 7–9 ปี[ 8 ]นอกจากนี้ เนื้องอก ALT/WDL ที่ถูกผ่าตัดออกไปแล้วอาจกลับมาเป็นซ้ำได้หลังจากช่วงเวลาที่แตกต่างกันไปในรูปแบบของมะเร็งไขมันชนิดที่เปลี่ยนแปลงสภาพ[ 6 ]การทดลองแบบสุ่มควบคุมขนาดใหญ่ที่เปรียบเทียบการฉายรังสีตามด้วยการผ่าตัดกับการผ่าตัดเพียงอย่างเดียวในเนื้องอก ALT/WDL พบว่ามีความแตกต่างกันเล็กน้อยระหว่างสองวิธีการรักษา การศึกษาขนาดเล็กที่ใช้สารยับยั้งแบบเลือกเฉพาะของผลิตภัณฑ์โปรตีนของ ยีน CDK4หรือMDM2ที่เกี่ยวข้องกับ ALT/WDL แสดงให้เห็นผลเพียงเล็กน้อยเท่านั้น การศึกษาเพิ่มเติมโดยใช้วิธีการรักษาเหล่านี้หรือวิธีการรักษาใหม่ทั้งหมดกำลังอยู่ระหว่างการตรวจสอบ[ 8 ]การศึกษาทบทวนในปี 2012 รายงานอัตราการรอดชีวิต 5 ปีและ 10 ปีของผู้ป่วยที่เป็น ALT/WDL อยู่ที่ 100% และ 87% ตามลำดับ[ 20 ]
การบำบัดแบบใหม่
วิธีการรักษาแบบใหม่ของ ALT/WDL นั้นเหมือนกับที่ระบุไว้ในหัวข้อ "วิธีการรักษาแบบใหม่" ของมะเร็งไขมันชนิดที่เซลล์มีการเปลี่ยนแปลงสภาพ (Dedifferentiated liposarcoma)
ลิโปซาร์โคมาที่เสื่อมสภาพ
ลิโปซาร์โคมาที่เสื่อมสภาพ (Dedifferentiated liposarcoma) เป็นเนื้องอกร้ายซึ่งในประมาณ 10% ของกรณีจะพัฒนามาจากเนื้องอกไขมันผิดปกติ/ลิโปซาร์โคมาที่แยกแยะได้ดี (ALT/WDL) ที่มีอยู่เดิม หรือที่บริเวณที่เคยผ่าตัดเอาเนื้องอก ALT/WDL ออกไป บุคคลที่ได้รับ การวินิจฉัยว่าเป็นเนื้องอกชนิดนี้ เป็นครั้งแรก อาจมี ALT/WDL ที่พัฒนาไปเป็นลิโปซาร์โคมาที่เสื่อมสภาพ แต่ตรวจไม่พบเนื่องจากพัฒนาโดยไม่มีอาการในบริเวณที่แยกตัวออกมามาก เช่น ช่องท้องส่วนหลังหรือช่องท้อง ลักษณะทางคลินิกและพันธุกรรมของลิโปซาร์โคมาที่เสื่อมสภาพหลายอย่างคล้ายคลึงกับที่พบในเนื้องอก ALT/WDL [ 8 ]
การนำเสนอ
ลิโปซาร์โคมาที่เสื่อมสภาพ (DDL) มักเกิดขึ้นในผู้ใหญ่วัยกลางคนและผู้สูงอายุ โดยมีอัตราการเกิดสูงสุดในช่วงอายุ 60-80 ปี[ 8 ]ในบางกรณี เนื้องอกเหล่านี้อาจพัฒนาในเด็กและวัยรุ่นได้[ 5 ]เนื้องอก DDL มักเกิดขึ้นในช่องว่างหลังเยื่อบุช่องท้อง แต่เช่นเดียวกับ ALT/WDL อาจเกิดขึ้นที่แขนขา บริเวณรอบอัณฑะ ช่องอก ศีรษะ หรือคอ[ 8 ]น้อยกว่า 1% ของ DDL ทั้งหมดพัฒนาในเนื้อเยื่ออ่อนผิวเผิน[ 8 ]หรือเบ้าตา[ 21 ]เมื่อตรวจพบครั้งแรก เนื้องอก DDL มักไม่เจ็บปวด มีขนาดใหญ่ อาจค่อยๆ ขยายใหญ่ขึ้นเรื่อยๆ เป็นเวลาหลายปี[ 8 ]และในการตรวจเอกซเรย์ ทั่วไป จะพบพื้นที่ที่มีการสะสมของแคลเซียม (ดังตัวอย่างในรูปที่ 1 ในส่วนพยาธิวิทยาของลิโปซาร์โคมา) [ 22 ] [ 23 ]ในบางกรณี ผู้ป่วยอาจมีอาการและ/หรือสัญญาณต่างๆ เนื่องจากการกดทับของเนื้องอกต่ออวัยวะ (เช่น อาการปวดท้องที่เกิดจากการอุดตันของลำไส้ หรือการอุดตันของทางเดินปัสสาวะที่เกิดจากการอุดตันของท่อปัสสาวะ ) ในบางกรณีที่พบได้น้อยมาก ผู้ป่วย DDL อาจมีอาการหรือสัญญาณของการอักเสบเรื้อรัง อย่างน้อยหนึ่งอย่าง (ดูอาการ B ) และ/หรือกลุ่มอาการพาราเนโอพลาสติก ที่เกี่ยวข้องกับต่อม ไร้ท่อระบบประสาทเยื่อเมือก ผิวหนังระบบเลือดหรือเนื้อเยื่ออื่นๆอาการและสัญญาณของการอักเสบเรื้อรังและกลุ่มอาการพาราเนโอพลาสติกต่างๆ เกิดจากการหลั่งของเนื้องอก เช่นไซโตไคน์ฮอร์โมนโปรสตาแกลนดินและ/หรือสารออกฤทธิ์ในระบบอื่นๆ ซึ่งจะหายไปอย่างสมบูรณ์หลังจากรักษา DDL ได้สำเร็จ[ 8 ]
พยาธิวิทยา
ลักษณะทางพยาธิวิทยาของเนื้องอก DDL (ดูรูปที่ 2 ในส่วนพยาธิวิทยาของไลโปซาร์โคมาด้านล่าง) มีความหลากหลายมาก แต่ส่วนใหญ่มักแสดงลักษณะของซาร์โคมาชนิดพลีโอโมฟิกที่ไม่จำแนกประเภท (ซึ่งเป็นเนื้องอกที่มีเซลล์ขนาดและรูปร่างแตกต่างกันจำนวนมาก และมีนิวเคลียส ขนาดและรูปร่างแตกต่างกัน ) หรือซาร์โคมาชนิดเซลล์รูปทรงกระสวย (ซึ่งเป็นเนื้องอกที่ประกอบด้วยเซลล์รูปทรงกระสวยใน พื้นหลัง ของเนื้อเยื่อเกี่ยวพัน ) ส่วนต่างๆ ของเนื้องอก DDL มักแสดงความแตกต่างในลักษณะของเนื้อเยื่อเกี่ยวพันพื้นหลัง: เนื้อเยื่อเหล่านี้อาจเป็นแบบไมซอยด์ (เช่น ประกอบด้วยสารใส คล้าย เมือกซึ่งเมื่อย้อมด้วย วิธี การย้อม H&E มาตรฐาน จะปรากฏเป็นสีน้ำเงินหรือม่วงมากกว่าสีแดงของเนื้อเยื่อปกติ) หรือไมโซคอลลาเจนัส (เช่น มีปริมาณเส้นใย คอ ลลาเจนสูง ในพื้นหลังไมซอยด์) และในประมาณ 5% ของกรณี จะมีบริเวณของออสทีออยด์ (ดูรูปที่ 1 ในส่วนพยาธิวิทยาของไลโปซาร์โคมาด้านล่าง) หรือวัสดุกระดูกอ่อน เนื้องอก ยังแสดงความแตกต่างอย่างมากในเนื้อหาของเซลล์ ตัวอย่างเช่น เนื้องอก DDL มากถึง 10% มีบริเวณที่มีพยาธิวิทยา ALT/WDL [ 8 ]และในกรณีที่หายากของ DDL จะมีบริเวณที่มีเซลล์แบนเรียงตัวเป็นวงคล้ายเยื่อหุ้มสมอง[ 24 ] [ 25 ]
พันธุศาสตร์
เซลล์มะเร็งในทั้ง DDL และ ALT/WDL มีโครโมโซมเครื่องหมายส่วนเกินขนาดเล็กที่ คล้ายกัน (sSMCs) และ/หรือโครโมโซมเครื่องหมายขนาดใหญ่ที่มีส่วนเพิ่มเติมของแขน q ของโครโมโซม 12 ที่แถบ 13 ถึง 15 บริเวณโครโมโซมนี้ประกอบด้วยยีนสองตัวที่เกี่ยวข้องกับการพัฒนาของเนื้องอก ได้แก่ ยีนMDM2 [ 26 ]และยีนCDK4 [ 27 ] [ 17 ]การมีสำเนาเพิ่มเติมของยีนทั้งสองนี้และ/หรือผลิตภัณฑ์โปรตีนที่ผลิตมากเกินไปเป็นตัวบ่งชี้ที่มีความไวและความจำเพาะสูงว่าเนื้องอกไขมันเป็น ALT/WDL หรือ DDL มากกว่าเนื้องอกไขมันชนิดอื่น[ 12 ] [ 17 ]การแสดงออกมากเกินไปของ ยีน MDM2และCDKและ/หรือวัสดุทางพันธุกรรมอื่น ๆ ใน sSMCs หรือโครโมโซมเครื่องหมายขนาดใหญ่คาดว่าจะส่งเสริมการพัฒนาและ/หรือความก้าวหน้าของเนื้องอก DDL เช่นเดียวกับเนื้องอก ALT/WDL [ 28 ]ยีนอื่นๆ ใน sMMC และโครโมโซมเครื่องหมายยักษ์ที่แสดงออกมากเกินไปในเซลล์มะเร็ง ALT/WDL และ DDL ได้แก่HMGA2 , CPM , YEATS4, [ 29 ]และDDIT3อย่างไรก็ตาม เมื่อเปรียบเทียบกับเซลล์มะเร็ง ALT/WDL เซลล์มะเร็ง DDL: 1)แสดงระดับยีนในโครโมโซมที่ผิดปกติทั้งสองในระดับที่สูงกว่า ซึ่งอาจมีส่วนทำให้ ALT/WDL พัฒนาไปเป็น DDL และ2)มีระดับผลิตภัณฑ์ยีนบนแขนยาวของโครโมโซม 1 ที่แถบ 32 แขนยาวของโครโมโซม 6 ที่แถบ 33 และในประมาณ 25% ของกรณี แขนสั้นของโครโมโซม 1 ที่แถบ 32.2 ซึ่งมี ยีน JUN อยู่ (ยีนนี้แสดงออกมากเกินไปใน DDL แต่ไม่ใช่ใน ALT/WDL) เนื่องจากผลิตภัณฑ์ของยีนJUN คือ c-junซึ่งยับยั้งการตายของเซลล์และส่งเสริมการเพิ่มจำนวนเซลล์ การผลิต c-jun มากเกินไปอาจส่งผลต่อความก้าวหน้าของ ALT/WDL ไปเป็น DDL และ/หรือความร้ายแรงของเซลล์มะเร็ง DDL [ 8 ]การวิเคราะห์การแสดงออกของยีน (เช่น การวัดการแสดงออกของผลิตภัณฑ์ของยีนหลายพันยีนที่สร้างโดยเซลล์ เนื้อเยื่อ หรือเนื้องอก) ได้เปิดเผยว่าการแยกตัวของเซลล์ ไขมัน และวิถีการเผาผลาญใน ALT/WDL มีการเพิ่มขึ้นในขณะที่การเพิ่มจำนวนเซลล์และ วิถี การตอบสนองต่อความเสียหายของ DNAมีการเพิ่มขึ้นใน DDL [ 6 ]
การวินิจฉัย
การตรวจทางพยาธิวิทยาของ DDL มักไม่ชัดเจนเพียงพอที่จะวินิจฉัยได้อย่างแน่ชัด อย่างไรก็ตาม การวินิจฉัย DDL ได้รับการสนับสนุนในบุคคลที่มีเนื้องอกประกอบด้วย ALT/WDL ผสมกับส่วนประกอบทางเนื้อเยื่อวิทยาของ DDL มีประวัติเคยเป็น ALT/WDL มาก่อน[ 8 ]หรือมีอาการของลิโปซาร์โคมาในช่องท้องส่วนหลัง (DDL คิดเป็นประมาณ 57% ของลิโปซาร์โคมาในช่องท้องส่วนหลังทั้งหมด) เนื้องอก DDL พบได้น้อยมาก (<1% ของกรณี) ในรูปของเนื้องอกผิวหนังชั้นนอก[ 8 ]มีโอกาสน้อยกว่า ALT/WDL เกือบ 5 เท่าที่จะเกิดขึ้นในเบ้าตา[ 14 ] [ 21 ]และพบได้น้อยมากในเด็ก[ 5 ]การตรวจพบการขยายตัวของ MDM2 ในเซลล์เนื้องอกถือเป็นมาตรฐานทองคำในการวินิจฉัยแยก WDL จากลิโปมา ลิโปมาผิดปกติ ซาร์โคมาเซลล์รูปทรงกระสวยที่ผิดปกติ ลิโปมาหลายรูปแบบ และเนื้องอกเส้นใยเดี่ยว[ 8 ] หรืออีกทางหนึ่ง การตรวจพบยีน CDK4ที่แสดงออกมากเกินไปในเซลล์เนื้องอกหรือการมีอยู่ของ sSMC ที่เกี่ยวข้องกับ ALT/WDL โดยเฉพาะ หรือโครโมโซมเครื่องหมายขนาดใหญ่ สนับสนุนการวินิจฉัย DDL หรือ ALT/WDL อย่างมาก[ 18 ] [ 19 ]ลักษณะทางคลินิก พยาธิวิทยา และความแตกต่างของยีน (เช่น การแสดงออกของ ยีน cJUN ในเซลล์เนื้องอก มากเกินไป สนับสนุนการวินิจฉัย DDL มากกว่า ATL/WDL อย่างมาก) ระหว่างลิโปซาร์โคมาสองรูปแบบหลังนี้ มักจะช่วยแยกแยะความแตกต่างระหว่างกันได้[ 8 ]
การรักษาและการพยากรณ์โรค
การผ่าตัดเอาเนื้องอกออกทั้งหมดมักเป็นวิธีการรักษาลำดับแรกที่แนะนำสำหรับเนื้องอก DDL ที่อยู่เฉพาะที่[ 6 ]อย่างไรก็ตาม การศึกษาที่กำลังเกิดขึ้นใหม่ชี้ให้เห็นว่าผู้ป่วยที่มีเนื้องอก DDL ที่จำกัดอยู่เฉพาะที่แขนขาหรือลำตัวและมีอัตราการรอดชีวิตโดยรวมที่เกี่ยวข้องกับเนื้องอกที่คาดการณ์ไว้ในระยะเวลา 10 ปี 51% หรือน้อยกว่านั้น จะมีผลลัพธ์ที่ดีขึ้นเมื่อเพิ่มเคมีบำบัด (เช่นโดโซรู บิซิน ร่วมกับไอโฟสฟาไมด์ ) เข้าไปในแผนการผ่าตัด [ 30 ]สำหรับ DDL ที่อยู่เฉพาะที่เหล่านี้อาจพิจารณาการฉายรังสี รอบการผ่าตัด ตาม แนวทางของ National Comprehensive Cancer Network ได้เช่นกัน [ 8 ]
DDL ในช่องท้องส่วนหลังเป็นรูปแบบของ DDL ที่พบได้บ่อยที่สุด เข้าถึงได้ยากทางการผ่าตัด และร้ายแรงที่สุด โดยมีอัตราการเกิดซ้ำ 66% และอัตราการรอดชีวิตโดยรวมใน 5 ปีอยู่ที่ 54% [ 31 ]ทางเลือกการรักษาหลักสำหรับ DDL ในช่องท้องส่วนหลังคือการผ่าตัดเอาเนื้องอกออกการทดลองทางคลินิกเฟส III พบว่าผลลัพธ์ของการรักษาด้วยรังสีตามด้วยการผ่าตัดเอาเนื้องอกออกนั้นมีความแตกต่างกันเล็กน้อยเมื่อเทียบกับการผ่าตัดเอาเนื้องอกออกเพียงอย่างเดียวในการรักษา DDL ในช่องท้องส่วนหลัง[ 6 ]ในการทดลองทางคลินิกเฟส III อื่นๆ ผู้ป่วย DDL ที่มีเนื้องอกในช่องท้องส่วนหลังที่เข้าถึงได้ยากและ/หรือเนื้องอกที่แพร่กระจายได้รับการรักษาด้วยเคมีบำบัดแบบแนวหน้า โดยเปรียบเทียบ doxorubicin กับ doxorubicin บวก ifosfamide หรือ doxorubicin กับgemcitabineบวกdocetaxelการศึกษาอื่นๆ ก็ได้ตรวจสอบคุณค่าของสูตรเคมีบำบัดต่างๆ เช่นกัน การศึกษาเหล่านี้มักพบว่ามีความแตกต่างกันเล็กน้อยในเวลาการรอดชีวิตโดยรวมในการเปรียบเทียบ แต่แสดงให้เห็นถึงการปรับปรุงบางอย่างในระยะเวลาการรอดชีวิตโดยปราศจากความคืบหน้าของโรคและพารามิเตอร์ทางคลินิกอื่นๆ จากการศึกษาเหล่านี้ การรักษาเบื้องต้นที่แนะนำสำหรับเนื้องอก DDL ในช่องท้องส่วนหลังและเนื้องอก DDL อื่นๆ ที่ไม่สามารถผ่าตัดได้หรือแพร่กระจาย คือการรักษาด้วยเคมีบำบัดแบบแอนทราไซคลิน หรือในกรณีที่เนื้องอกดื้อต่อการรักษาหรือกลับมาเป็นซ้ำ ให้ ใช้เคมีบำบัด อีริบูลินการทบทวนที่ดำเนินการในปี 2020 รายงานว่าระยะเวลาการอยู่รอดเฉลี่ยสำหรับ DDL ที่มีระดับทางพยาธิวิทยาต่ำและระดับสูงคือ 113 เดือนและ 48 เดือนตามลำดับ[ 32 ]จำเป็นต้องมีการศึกษาเพิ่มเติมเพื่อให้ได้หลักฐานเกี่ยวกับประสิทธิภาพของการฉายรังสี เคมีบำบัด และการรักษาแบบใหม่ใน DDL ทุกประเภท[ 33 ]
การบำบัดแบบใหม่
ปัจจุบันมี การทดลองทางคลินิกเกี่ยวกับสูตรการรักษาใหม่หลายวิธีสำหรับ DDL และกรณี ALT/WDL ที่รุนแรงหรือมีปัญหาอื่นๆการศึกษาทางคลินิกเฟส II ที่กำลังตรวจสอบabemaciclib กำลังดำเนินการอยู่ในผู้ป่วย DDL ที่ได้รับการรักษามาก่อนหรือไม่ได้รับการรักษา การวิเคราะห์เบื้องต้นแสดงให้เห็นว่าสารยับยั้ง เอนไซม์ Serine/threonine-specific protein kinase ซึ่งเป็น ผลิตภัณฑ์ของยีนCDK4และCDK6 นี้ ทำให้ระยะเวลาการรอดชีวิตโดยปราศจากความคืบหน้าของโรคโดยเฉลี่ยยาวนานขึ้นถึง 30.4 สัปดาห์[ 6 ] การศึกษาทางคลินิก เฟสIII แบบหลายศูนย์ แบบสุ่ม แบบตาบอดสองทาง และ ควบคุมด้วย ยาหลอกของ abemaciclib กำลังอยู่ในขั้นตอนการดำเนินการ และจะเริ่มรับสมัครผู้ป่วย 108 รายที่มี DDL ขั้นสูง กำเริบซ้ำ และ/หรือแพร่กระจายในเร็วๆ นี้ (ตามที่ระบุในเดือนกรกฎาคม 2021) การศึกษานี้ได้รับการสนับสนุนโดย Sarcoma Alliance for Research through Collaboration [ 34 ]ร่วมกับEli Lilly and Company [ 35 ] Ribociclib ซึ่งเป็นสารยับยั้งยีน CDK4และCDK6เช่นกันเมื่อใช้ร่วมกับสารยับยั้งmTOR อย่าง everolimusอยู่ในขั้นตอนการทดลองทางคลินิกเฟส II ในผู้ป่วย DDL หรือ leiomyosarcoma ขั้นสูง[ 35 ]การศึกษาเพื่อขึ้นทะเบียนเฟส III (เช่น การศึกษาเพื่อยืนยันขนาดใหญ่ที่มุ่งสร้างโปรไฟล์ผลประโยชน์/ความปลอดภัยที่ยอมรับได้ เพื่อให้ได้รับการอนุมัติจากหน่วยงานกำกับดูแลสำหรับข้อบ่งชี้ที่กำหนดไว้อย่างแม่นยำ) กำลังประเมินความปลอดภัยและประสิทธิภาพของmilademetanเมื่อเทียบกับ trabectedin ในผู้ป่วยที่มี DDL ที่ไม่สามารถผ่าตัดได้ (เช่น การผ่าตัดถือว่าทำให้เกิดความเจ็บป่วยหรือเสียชีวิตที่ไม่สามารถยอมรับได้) หรือ DDL ที่แพร่กระจายซึ่งมีความก้าวหน้าหลังจากได้รับการรักษาด้วยระบบอย่างน้อย 1 ครั้ง รวมถึงการรักษาด้วย anthracycline อย่างน้อย 1 ครั้ง ผู้สนับสนุน Rain Therapeutics Inc กำลังรับสมัครผู้เข้าร่วมการทดลอง 160 คน[ 36 ]การทดลองทางคลินิกเฟส III อีกครั้งหนึ่งกำลังตรวจสอบ สารยับยั้ง MDM2 milademetan [ 37 ]เทียบกับtrabectedin ซึ่งเป็นตัวบล็อกของปัจจัยการถอดรหัส ที่ก่อให้เกิดมะเร็ง FUS-CHOPในALT/WDL และ DDL ที่มีการแสดงออกของMDM2 มากเกินไป [ 38 ] Milademetan แสดงให้เห็นถึงความเป็นพิษที่จัดการได้และกิจกรรมบางอย่างที่ส่งผลให้โรคคงที่และ/หรือมีการตอบสนองบางส่วนใน DDL [ 6 ]
มะเร็งไขมันชนิดมิกซอยด์
การนำเสนอ
ลิโปซาร์โคมาชนิดไมซอยด์ (MLS) ซึ่งรวมถึงลิโปซาร์โคมาชนิดหนึ่งที่เรียกว่าลิโปซาร์โคมาเซลล์กลม[ 39 ]คิดเป็นประมาณ 30% ของลิโปซาร์โคมาทั้งหมด มีอัตราการเกิดสูงสุดในบุคคลอายุ 40-50 ปี โดยพบในเพศชายมากกว่าเพศหญิงในงานวิจัยส่วนใหญ่ แม้ว่าจะไม่พบบ่อยในเด็กและวัยรุ่น แต่ MLS เป็นลิโปซาร์โคมาชนิดที่พบได้บ่อยที่สุดในกลุ่มอายุเหล่านี้ โดยทั่วไป MLS จะปรากฏเป็นก้อนขนาดใหญ่ (1 ถึง 39 ซม. เฉลี่ย 12 ซม.) เคลื่อนที่ได้ มีขอบเขตชัดเจน ไม่เจ็บปวด ซึ่งพัฒนาขึ้นตั้งแต่ 1 สัปดาห์ถึง 15 ปีก่อนการวินิจฉัย เนื้องอก MLS ตั้งอยู่ในเนื้อเยื่ออ่อนที่อยู่ลึกของต้นขา (65–80% ของกรณี) ขาส่วนล่าง (10–15% ของกรณี) ช่องท้องส่วนหลัง (8% ของกรณี) และแขน (5% ของกรณี) ในประมาณหนึ่งในสามของกรณี เนื้องอกเหล่านี้จะแพร่กระจายไปยังเนื้อเยื่ออ่อนอื่นๆ (เช่น ช่องท้องส่วนหลัง ช่องอก หรือแขนขาอื่นๆ) กระดูก และ/หรือปอด ผู้ป่วยอาจมีอาการแสดงของการแพร่กระจายเหล่านี้ โดยเฉพาะอย่างยิ่งในกระดูก มีคำแนะนำว่าผู้ป่วยควรได้รับการตรวจหาการแพร่กระจายของกระดูกเมื่อมาพบแพทย์โดยใช้ภาพทางการแพทย์รวมถึง การ เอกซเรย์การสแกน CTและ/หรือ การถ่าย ภาพด้วยคลื่นแม่เหล็กไฟฟ้า[ 40 ]
พยาธิวิทยา
การวิเคราะห์ทางพยาธิวิทยาของ MLS (ดูรูปที่ 3 และ 4 ในส่วนพยาธิวิทยาของลิโปซาร์โคมาด้านล่าง) เผยให้เห็นเซลล์ที่กระจายอยู่ทั่วเมทริกซ์เมือก (เช่น พื้นหลังของเนื้อเยื่อเกี่ยวพันที่ปรากฏเป็นสีน้ำเงินหรือสีม่วงมากกว่าสีแดงของเนื้อเยื่อเกี่ยวพันปกติเมื่อเนื้อเยื่อเหล่านี้ได้รับการเตรียมอย่างเหมาะสมย้อมด้วย H&Eและดูด้วยกล้องจุลทรรศน์) เซลล์เหล่านี้คือลิโปบลาสต์ซึ่งบางส่วนมีรูปร่างคล้ายแหวนตราประทับ (รูปร่างที่บ่งชี้ว่าเซลล์อาจเป็นเนื้องอก) รูปไข่ หรือรูปทรงกลม[ 40 ]เนื้องอก MLS อาจมีเซลล์หนาแน่นและมีแผ่นเซลล์กลมทึบซึ่งประกอบด้วยเซลล์อย่างน้อย 5% ของเซลล์ทั้งหมด หรือมีเซลล์น้อยซึ่งมีนิวเคลียสเรียบและมีเซลล์กลม <5% ในพื้นหลังของเส้นเลือดฝอยโค้งงอคล้ายกับลวดลายตาข่ายลวด เนื้องอกที่มีเซลล์กลมอย่างน้อย 5% จัดเป็นเนื้องอกเกรดสูง ในขณะที่เนื้องอกที่มีเซลล์กลม <5% จัดเป็นเนื้องอกเกรดต่ำ[ 39 ]เนื้องอก MLS ระดับสูงมักจะมีอาการทางคลินิกที่รุนแรงกว่าเนื้องอก MLS ระดับต่ำ[ 40 ]
พันธุศาสตร์
เซลล์เนื้องอก MLS แทบจะถูกกำหนดโดยการแสดงออกของยีนฟิวชั่นFUS-DDIT3 (เรียกอีกอย่างว่ายีนไคเมอริก ) ซึ่งเกิดขึ้นใน >95% ของกรณี หรือ ยีนฟิวชั่น EWSR1-DDIT3ซึ่งเกิดขึ้นใน <5% ของกรณีที่เหลือ ยีนฟิวชั่น FUS-DDIT3เกิดขึ้นจากการย้ายตำแหน่ง (เรียกว่า t(12:16)(q13:p11)) ระหว่างตำแหน่งของยีนDDIT3 [ 41 ]ที่แถบ 12 ของแขน q ของโครโมโซม 12 และตำแหน่งของยีนFUS [ 42 ]ที่แถบ 11 บนแขนสั้นของโครโมโซม 16 (เรียกอีกอย่างว่าแขนp ) ผลิตภัณฑ์ โปรตีนฟิวชั่น (เรียกอีกอย่างว่าโปรตีนไคเมอริก) ของยีน ออนโคยีนไคเมอริก FUS-DDIT3 นี้เป็นที่ทราบกันว่าสามารถยับยั้งการเจริญเติบโตของเซลล์ไขมันและส่งเสริมการเกิดเนื้องอกได้ ยีน ฟิวชั่น EWSR1-DDIT3 (เรียกว่า t(12;22)(q13;q12)) เกิดจากการย้ายตำแหน่งของยีนEWSR1 [ 43 ]ซึ่งอยู่ที่แถบ 12.2 บนแขน q ของโครโมโซม 22 พร้อมกับ ยีน DDIT2ผลิตภัณฑ์โปรตีนฟิวชั่นของ ยีน EWSR1-DDIT3เช่นเดียวกับโปรตีนฟิวชั่น FUS-DDIT3 ส่งเสริมการเกิดเนื้องอก[ 44 ]แม้ว่าจะมีความสัมพันธ์ของยีนฟิวชั่นเหล่านี้ แต่ก็ยังจำเป็นต้องมีการศึกษาเพิ่มเติมเพื่อกำหนดบทบาทของยีนเหล่านี้ต่อการพัฒนาและ/หรือการคงอยู่ของเนื้องอก MLS [ 6 ]
การวินิจฉัย
เนื้องอก MLS ระดับต่ำและระดับกลางสามารถระบุได้ทางเนื้อเยื่อวิทยาโดยลักษณะทางสัณฐานวิทยาแบบคลาสสิกของหลอดเลือดที่มีลักษณะคล้ายตาข่ายไก่ที่กระจายอยู่ทั่วเนื้อเยื่อเกี่ยวพันแบบเมือก อย่างไรก็ตาม เนื้องอก MLS ระดับสูงอาจแยกแยะได้ยากจากเนื้องอกเซลล์กลมชนิดอื่น โดยเฉพาะอย่างยิ่งเนื้องอก MLS ระดับสูงที่ประกอบด้วยเซลล์กระจายและ/หรือเซลล์กลมบริสุทธิ์ในระดับที่บดบังรูปแบบหลอดเลือด-เมือกแบบคลาสสิกนี้ การตรวจพบ การจัดเรียงยีน DDIT3ใหม่ร่วมกับ ยีน FUSหรือEWSR1โดยการผสมแบบ in situหรืออิมมูโนฮิสโตเคมีหรือ การถอดรหัสฟิวชั่น RNAของยีนเหล่านี้โดยปฏิกิริยาลูกโซ่พอลิเมอเรสแบบเรียลไทม์ยืนยันการวินิจฉัยเนื้องอก MLS ระดับสูง เช่นเดียวกับกรณีที่คลุมเครือของเนื้องอก MLS ระดับต่ำหรือระดับกลาง[ 44 ]
การรักษาและการพยากรณ์โรค
โดยทั่วไป MLS จะได้รับการรักษาด้วยการผ่าตัด แต่บางครั้งอาจต้องมีการแทรกแซงที่รุนแรงกว่า เช่น การตัดแขนขาอาจจำเป็นเมื่อ เส้นประสาท และหลอดเลือด ของแขนขา ได้รับความเสียหาย ความเสี่ยงของการเกิดซ้ำหลังการผ่าตัดภายใน 3 ปีหลังการผ่าตัดมีรายงานว่าอยู่ที่ประมาณ 15% เมื่อไม่ได้เอาเนื้องอกออกทั้งหมด และประมาณ 10% เมื่อเอาเนื้องอกออกทั้งหมด[ 40 ]การเพิ่มการฉายรังสีร่วมกับการผ่าตัดได้ช่วยควบคุมเนื้องอก MLS ในบริเวณนั้นได้ดีขึ้น และได้รับการแนะนำให้ใช้ในการรักษา MLS ที่ไม่สามารถผ่าตัดได้และ MLS ที่กลับมาเป็นซ้ำ[ 45 ]อย่างไรก็ตาม จำเป็นต้องมีการศึกษาเพิ่มเติมเพื่อกำหนดคุณค่าของการฉายรังสีในการรักษา MLS ชนิดต่างๆ[ 40 ]พบว่าสูตรเคมีบำบัดที่ใช้ifosfamide , anthracycline เช่น daunorubicin , dacarbazineและ/หรือtrabectedin มีประโยชน์: การทดลองทางคลินิกเฟส III แสดงให้เห็นว่าระยะเวลาการอยู่รอดโดยปราศจากความคืบหน้าของโรคในผู้ป่วย MLS ที่ได้รับการรักษาด้วย trabectedin หรือ dacarbazine คือ 5.6 และ 1.5 เดือน ตามลำดับ ในปี 2558 องค์การอาหารและยาได้อนุมัติให้ใช้ trabectedin ในการรักษา liposarcoma ที่ไม่สามารถผ่าตัดได้และแพร่กระจาย
โดยรวมแล้ว อัตราการรอดชีวิต 10 ปีของผู้ป่วย MLS อยู่ที่ 77% ซึ่งเป็นอัตราการรอดชีวิตที่ยาวนานกว่าลิโปซาร์โคมาชนิดอื่นๆ อย่างเห็นได้ชัด เมื่อเทียบกับ MLS ที่มีความเสี่ยงต่ำ MLS ที่มีความเสี่ยงสูง (ความเสี่ยงที่กำหนดโดยปริมาณเซลล์กลมในเนื้องอกและ/หรือตัวบ่งชี้การพยากรณ์โรคที่ไม่ดีอื่นๆ) จะสัมพันธ์กับอัตราการแพร่กระจายที่เพิ่มขึ้น และด้วยเหตุนี้จึงมีระยะเวลาการรอดชีวิตที่สั้นลง ขนาดเนื้องอกที่ใหญ่ขึ้น (≥ 10 ซม.) สัมพันธ์อย่างมากกับ MLS ที่มีเกรดสูงขึ้น และด้วยเหตุนี้จึงมีระยะเวลาการรอดชีวิตที่สั้นลง ปัจจัยอื่นๆ ที่เกี่ยวข้องกับผลลัพธ์ที่ไม่ดีใน MLS ได้แก่ การมีเนื้อตายของเนื้องอก อายุ >45 ปีการแสดงออกของยีนP53 มากเกินไป [ 40 ]และเพศชาย[ 46 ]ลิโปซาร์โคมาชนิดเซลล์กลมก็ดูเหมือนจะมีพยากรณ์โรคที่ไม่ดีนักเช่นกัน: ในการทบทวนย้อนหลังต่างๆ พบว่าลิโปซาร์โคมาชนิดเซลล์กลมมักจะเป็นเกรดต่ำและตอบสนองต่อเคมีบำบัดได้ค่อนข้างดี ในขณะที่ลิโปซาร์โคมาชนิดเซลล์กลมเกรดสูงมีอัตราการแพร่กระจายสูงกว่า มีพฤติกรรมที่รุนแรงกว่า และไม่ตอบสนองต่อเคมีบำบัดได้ดี[ 40 ]อย่างไรก็ตาม สิ่งสำคัญที่ควรทราบคือ ลิโปซาร์โคมาชนิดเซลล์กลมในผู้ป่วยเด็กเกือบทุกรายมีพยากรณ์โรคที่ดีเยี่ยม[ 45 ]
การบำบัดแบบใหม่
อีฟาตูทาโซน (efatutazone) ซึ่งเป็นตัวกระตุ้นPPAR-γ [ 47 ]ได้รับการศึกษาในการทดลองระยะที่ 1 ขนาดเล็กในผู้ป่วยที่มีมะเร็งระยะลุกลามหลายชนิด ยานี้ทำให้เกิดการตอบสนองที่คงทนอย่างเห็นได้ชัดในผู้ป่วย MLS ซึ่งบ่งชี้ว่าตัวกระตุ้น PPAR-γ จะมีประโยชน์ในการรักษาโรคนี้[ 48 ]การทดลองทางคลินิกระยะที่ 2 ที่ดำเนินการในอิตาลีกำลังตรวจสอบผลของทราเบคทีดินร่วมกับพิโอไกลทาโซน (ตัวกระตุ้น PPAR-γ อีกตัวหนึ่ง) ในผู้ป่วยที่มีเนื้องอก MLS ที่คงที่ การศึกษานี้ประกอบด้วยสองขั้นตอนต่อเนื่องกัน ขั้นตอนแรกจะตรวจสอบการตอบสนองของผู้ป่วยที่ได้รับการรักษาด้วยทราเบคทีดินเพียงอย่างเดียวอย่างน้อย 4 รอบ หากโรคคงที่ ขั้นตอนที่สองจะตรวจสอบผลของการรักษาผู้ป่วยที่ตอบสนองในเบื้องต้นด้วยการใช้ทราเบคทีดินและพิโอไกลทาโซนร่วมกัน[ 49 ]การทดลองทางคลินิกระยะที่ 2 ใกล้จะเสร็จสมบูรณ์เพื่อประเมินประสิทธิภาพของไซโรลิมัส (สารยับยั้งMTOR ; ไซโรลิมัสยังเป็นที่รู้จักในชื่อราพาไมซิน) ร่วมกับไซโคลฟอสฟาไมด์ (ยาเคมีบำบัด) ใน MLS ที่แพร่กระจายหรือผ่าตัดไม่ได้[ 50 ]การทดลองทางคลินิกระยะที่ 2 กำลังรับสมัครผู้ป่วยเพื่อประเมินซินทิลิแมบ ( แอนติบอดีโมโนโคลนอลIgG4 ของมนุษย์ ที่มุ่งเป้าไปที่โปรตีนการตายของเซลล์แบบโปรแกรม 1ที่อยู่บนพื้นผิวของเซลล์) ร่วมกับยาเคมีบำบัด 2 ชนิด ได้แก่ด็อกโซรูบิซินและไอโฟสฟาไมด์เป็นการรักษาเบื้องต้นของมะเร็งเนื้อเยื่ออ่อนรวมถึง MLS [ 51 ]
เซลล์ Tได้รับการดัดแปลงทางพันธุกรรมเพื่อกำหนดเป้าหมายแอนติเจนMAGE-A4 ที่แสดงออกบน เปปไทด์ที่มี MAGE-A4 ของ HLA-A*02ซึ่งอยู่บนพื้นผิวของเซลล์มะเร็งในเนื้องอกบางชนิด เซลล์ที่ได้รับการดัดแปลงเหล่านี้ (เรียกว่าเซลล์ ADP-A2M4-T) โจมตีและฆ่าเซลล์มะเร็งของมนุษย์ที่เพาะเลี้ยง หลายชนิดที่มีแอนติเจนนี้ [ 52 ]และในการศึกษาทางคลินิกระยะที่ 1 พบว่าทำให้เนื้องอกแข็งชนิดต่างๆ หดตัวลงในผู้ป่วยที่มีเนื้องอกซึ่งมีเซลล์มะเร็งที่แสดงออกแอนติเจนนี้[ 53 ]การศึกษาทางคลินิกระยะที่ 2 ได้คัดเลือกบุคคลเพื่อตรวจสอบประสิทธิภาพและความปลอดภัยของเซลล์ ADP-A2M4 T (ที่ได้รับการดัดแปลงจากเซลล์ T ของผู้รับเอง) ในผู้ป่วยที่มี HLA-A*02 เป็นบวกที่มีเนื้องอก MLS ระยะลุกลามหรือผ่าตัดไม่ได้และมี MSGE-4 เป็นบวก[ 54 ]
ลิโปซาร์โคมาชนิดพลีโอโมฟิก
การนำเสนอ
ลิโปซาร์โคมาชนิดเพลโอโมฟิก (PLS) ซึ่งคิดเป็น 5% ถึง 10% ของกรณีลิโปซาร์โคมาทั้งหมด[ 55 ]เป็นเนื้องอกของเซลล์ไขมันที่เติบโตเร็ว มักมีขนาดใหญ่ (>5 ซม.) และไม่เจ็บปวด แต่มีความร้ายแรงสูง [ 56 ] มักเกิดขึ้นในผู้ที่มีอายุมากกว่า 50 ปี[ 56 ]และพบในเพศหญิงมากกว่า[ 45 ]เนื้องอก PLS พบได้น้อยในเด็ก[ 56 ]เนื้องอก PLS มักพบที่ขาหรือแขน (65% ของกรณี) ช่องท้องส่วนหลังหรือช่องท้อง (15% ของกรณี) [ 6 ] หรือในกรณีที่หายาก อาจพบที่ผนังลำตัวท่ออสุจิ [ 56 ]บริเวณศีรษะและลำคอ[ 57 ]ผนังทรวงอกช่องเชิงกรานเยื่อหุ้มปอด เยื่อหุ้มหัวใจและกระดูกสันหลัง[ 55 ]เนื้องอกเหล่านี้มักจะอยู่ในเนื้อเยื่ออ่อนส่วนลึก โดยมีเพียง 25% ของกรณีเท่านั้นที่พบในเนื้อเยื่อใต้ผิวหนัง[ 56 ]กรณีหายากของ PLS เกิดขึ้นในบุคคลที่มี กลุ่มอาการ Li-FraumeniหรือMuir–Torre ซึ่ง เป็นความผิดปกติทางพันธุกรรมสองชนิดที่ทำให้ผู้ที่ได้รับผลกระทบมีแนวโน้มที่จะเป็นมะเร็งชนิดต่างๆ[ 45 ]
พยาธิวิทยา
พยาธิวิทยาของเนื้องอก PLS มักประกอบด้วยบริเวณที่มีลักษณะคล้ายไมซอยด์ไลโปซาร์โคมา[ 58 ]ผสมกับบริเวณที่มีเซลล์ที่ยังไม่แตกต่าง[ 56 ]เนื้องอกเหล่านี้มีเซลล์หนาแน่นมากและมีไลโปบลาสต์ รูปร่างต่างๆ อย่างน้อยบางส่วน ที่มีนิวเคลียสหลายรูปแบบ[ 58 ]บริเวณเนื้อตายพบได้บ่อย เซลล์ยักษ์ ซึ่งบางส่วนมีหลายนิวเคลียสและ/หรือมีนิวโทรฟิลที่ถูกกลืนกินพบได้เป็นครั้งคราว และ อาจพบหยด ไฮยาลีนในบางเซลล์ รวมถึงกระจายอยู่นอกเซลล์ทั่วทั้งเนื้องอก[ 59 ]ส่วนประกอบที่ยังไม่แตกต่างของเนื้องอกเหล่านี้ส่วนใหญ่มักประกอบด้วยเซลล์รูปทรงกระสวย โดย 25% ของกรณีแสดงเซลล์ที่มีสัณฐานวิทยา ของ เซลล์เยื่อบุผิวเนื้องอกเหล่านี้มีจุดอย่างน้อยบางจุดที่มีพยาธิวิทยาคล้ายกับฮิสติโอไซโตมาชนิดไมโซไฟโบรซาร์โคมา เกรดสูง [ 56 ] [ 58 ] [ 60 ] ซึ่งเป็นเนื้องอกที่เคยเรียกว่ามะเร็งไมซอยด์ไฟบรอยด์ฮิสติโอไซโตมา[ 61 ]
พันธุศาสตร์
เซลล์มะเร็ง PLS มีความผิดปกติของยีนและโครโมโซมหลายอย่าง: ยีนTP53 ถูก ลบหรือกลายพันธุ์ใน 17–60% ของกรณี; ยีน RB1ถูกลบใน 60% ของกรณี; และ ยีน Neurofibromin 1หายไปจากการกลายพันธุ์ ที่ทำให้ไม่ทำงาน ใน 8% ของกรณี หรือในกรณีที่หายากกว่านั้นคือการลบบริเวณตำแหน่งในแถบ 11.2 บนแขนยาวของโครโมโซม 12 เซลล์เหล่านี้ยังอาจแสดงการเพิ่มขึ้นของสารพันธุกรรมบริเวณ: แถบ 12–15 บนแขนสั้นของโครโมโซม 5; แถบ 21 บนแขนสั้นของโครโมโซม 1; และแถบ 22 บนแขนยาวของโครโมโซม 7 การเปลี่ยนแปลงจำนวนสำเนาของยีนที่เกิดจากความผิดปกติเหล่านี้คล้ายกับที่พบในฮิสติโอไซโตมาชนิดไมโซไฟโบรซาร์โคมาบทบาทของการเปลี่ยนแปลงจำนวนสำเนาของยีนเหล่านี้ในการส่งเสริม PLS ยังไม่ได้รับการกำหนด ดังนั้น PLS จึงแตกต่างจากลิโปซาร์โคมาชนิดอื่นตรงที่เซลล์มะเร็งมีจีโนมที่ซับซ้อนโดยไม่มีการเปลี่ยนแปลงทางจีโนมที่เป็นลักษณะเฉพาะหรือยีนที่สามารถระบุได้ซึ่งเป็นตัวขับเคลื่อนโรค การตรวจพบการเปลี่ยนแปลงในการแสดงออกของ ยีน TP53, RB1และneurofibromin 1รวมถึงยีนอื่นๆ ที่มีการเปลี่ยนแปลงน้อยกว่าใน PLS (เช่นPIK3CA , ไทโรซีนโปรตีนไคเนส SYK , PTK2B , EPHA5และERBB4 ) อาจช่วยสนับสนุนแต่ไม่ได้ระบุอย่างชัดเจนว่าเนื้องอกนั้นเป็น PLS [ 6 ] [ 56 ] การขยายปลาย เทโลเมียร์ ของโครโมโซมโดยกลไกทางพยาธิวิทยาที่เรียกว่าการยืดเทโลเมียร์แบบทางเลือกเกิดขึ้นในเซลล์มะเร็งของ PLS ประมาณ 80% แต่พบได้น้อยกว่ามากหรือไม่พบในลิโปซาร์โคมาอีกสี่รูปแบบ[ 58 ]
การวินิจฉัย
การวินิจฉัย PLS ขึ้นอยู่กับลักษณะอาการ พยาธิวิทยา และพันธุกรรม พยาธิวิทยาของ PLS มักจะคล้ายคลึงกับ myxofibrosarcoma แต่แตกต่างจากเนื้องอกชนิดนั้นตรงที่มี lipoblasts ที่มีรูปร่างหลากหลาย[ 58 ]
การรักษาและการพยากรณ์โรค
การผ่าตัดเอาส่วนที่เป็นโรคออกทั้งหมดเป็นวิธีการรักษาหลักสำหรับ PLS นอกจากนี้ยังเป็นการรักษาแบบประคับประคองที่สำคัญเพื่อบรรเทาอาการเนื่องจากการกดทับของอวัยวะและเนื้อเยื่อ การผ่าตัดอาจต้องเอาอวัยวะที่ถูกกดทับออกทั้งหมด เช่น ไตหรือลำไส้ใหญ่ อย่างไรก็ตาม แม้จะมีการผ่าตัดนี้ อัตราการกลับมาเป็นซ้ำในบริเวณเดิมก็ยังสูงมาก การใช้เคมีบำบัดและ/หรือรังสีบำบัดร่วมกับการผ่าตัดเอาส่วนที่เป็นโรคออกทั้งหมดไม่ได้แสดงให้เห็นว่าช่วยยืดอายุการรอดชีวิตและถือเป็นการรักษาที่ยังเป็นที่ถกเถียงกันอยู่[ 55 ]เครือข่ายมะเร็งแห่งชาติแนะนำให้รักษาผู้ป่วยที่มี PLS เฉพาะที่ที่มีความเสี่ยงสูงด้วยการผ่าตัดเอาส่วนที่เป็นโรคออกทั้งหมดเมื่อทำได้ ร่วมกับการฉายรังสี ผู้ป่วยที่มีโรคแพร่กระจายได้รับการรักษาด้วยเคมีบำบัด (เช่นโดโซรูบิ ซิน ร่วมกับ ไอโฟ สฟาไมด์หรืออีริบูลิน ) คล้ายกับสูตรที่ใช้สำหรับลิโปซาร์โคมาที่เสื่อมสภาพ (ดูส่วนด้านบนเกี่ยวกับการรักษาลิโปซาร์โคมาชนิดนี้) [ 6 ]ประมาณ 20% ของเนื้องอก PLS แพร่กระจายไปยังอวัยวะที่อยู่ห่างไกล ซึ่งที่พบได้บ่อยที่สุดคือปอด (82% ของการแพร่กระจาย) ตับ (18% ของการแพร่กระจาย) และกระดูกหรือตับอ่อน (18% ของการแพร่กระจาย) อัตราการรอดชีวิตของ PLS ที่ 1, 3 และ 5 ปี มีรายงานอยู่ที่ 93%, 75% และ 29% ตามลำดับ เนื้องอกที่อยู่ตรงกลางลำตัว มีขนาดใหญ่กว่า 10 ซม. อยู่ลึก หรือมีบริเวณเนื้อตาย มีพยากรณ์โรคที่แย่กว่า[ 55 ]
ลิโปซาร์โคมาชนิดเมือกหลายรูปแบบ
ลิโปซาร์โคมาชนิดไมซอยด์พลีโอโมฟิก (เดิมเรียกว่าลิโปซาร์โคมาชนิดไมซอยด์พลีโอโมฟิก[ 62 ] ) ได้รับการอธิบายครั้งแรกในการศึกษาลิโปซาร์โคมาขนาดใหญ่ในปี 2009 [ 63 ]แม้ว่าในตอนแรกจะถือว่าเป็นรูปแบบหนึ่งของลิโปซาร์โคมาชนิดไมซอยด์ที่มีลักษณะพลีโอโมฟิกแต่องค์การอนามัยโลก (2020) ได้จัดประเภทให้เป็นลิโปซาร์โคมาชนิดใหม่และแตกต่างออกไป การจัดประเภทนี้ขึ้นอยู่กับการค้นพบว่าลิโปซาร์โคมาชนิดไมซอยด์พลีโอโมฟิก แม้จะมีลักษณะทางพยาธิวิทยาที่คล้ายกับลิโปซาร์โคมาชนิดไมซอยด์ แต่ก็มีลักษณะทางคลินิก และที่สำคัญที่สุดคือลักษณะทางพันธุกรรมและโมเลกุลที่สำคัญที่แตกต่างจากลิโปซาร์โคมาชนิดไมซอยด์และลิโปซาร์โคมาอีกสามรูปแบบ[ 5 ]
การนำเสนอ
มะเร็งไขมันชนิด Myxoid pleomorphic liposarcoma (MPL) เป็นมะเร็งไขมันชนิดหนึ่งที่หายากและรุนแรงมาก ซึ่งมักเกิดขึ้นในเด็ก วัยรุ่น[ 5 ]ผู้ใหญ่ตอนต้น[ 6 ]และจากการศึกษาล่าสุดพบว่าเกิดขึ้นในบุคคลที่มีอายุมากกว่า 50 ปี[ 62 ]เนื้องอก MPL มักปรากฏเป็นก้อนเนื้อเยื่ออ่อนที่อยู่ลึก ซึ่งมักพบในช่องอก[ 44 ]และพบได้น้อยในแขนขา ศีรษะและคอ ช่องท้อง หรือลำตัว[ 6 ]อย่างน้อยสองกรณีของ MPL พบในบุคคลที่เป็นโรค Li–Fraumeniซึ่งเป็นโรคทางพันธุกรรมที่ทำให้บุคคลมีแนวโน้มที่จะเป็นมะเร็งชนิดต่างๆ[ 58 ] [ 64 ] [ 65 ]
พยาธิวิทยา
จากการวิเคราะห์ทางพยาธิวิทยา พบว่าเนื้องอก MPL ประกอบด้วยบริเวณที่มีลักษณะคล้ายกับไลโปซาร์โคมาชนิดไมซอยด์ทั่วไป บริเวณเหล่านี้ซึ่งคิดเป็น 30–50% ของพื้นที่เนื้องอกทั้งหมด มีเมทริกซ์ไมซอยด์จำนวนมาก มีหลอดเลือดฝอยที่พัฒนาอย่างดี มีเซลล์ที่ไม่รุนแรง มีรูปร่างกลมและ/หรือเป็นรูปทรงกระสวยเล็กน้อย มีลิโปบลาสต์ที่มีช่องว่าง และมีเซลล์หลายนิวเคลียสรูปร่างคล้ายดอกไม้ขนาดเล็ก อย่างไรก็ตาม บริเวณเหล่านี้ยังมีเซลล์ที่มีรูปร่างผิดปกติกระจายอยู่ทั่วไป ซึ่งแสดงให้เห็นถึงการขยายตัวของนิวเคลียสและความไม่สม่ำเสมอมากกว่าเซลล์ที่พบในเนื้องอกไลโปซาร์โคมาชนิดไมซอยด์ บริเวณอื่นๆ ของเนื้องอก MPL มีเซลล์มากกว่าและประกอบด้วยลิโปบลาสต์ที่ เติบโตอย่างรวดเร็วและมีรูปร่างผิดปกติสูง [ 62 ]
พันธุศาสตร์
เซลล์มะเร็งใน MPL ไม่แสดง ยีนฟิวชั่น FUS-DDIT3หรือEWSR1-DDIT3ซึ่งแสดงโดยเซลล์มะเร็งในกรณี myxoid fibrosarcoma มากกว่า 95% หรือน้อยกว่า 5% ตามลำดับ[ 62 ] [ 6 ]การทำงานของยีนยับยั้งเนื้องอกRB1 ไม่ทำงาน เนื่องจากการลบหรือการยับยั้งทางพยาธิวิทยาพบได้ในทุกกรณีของ MPL เซลล์มะเร็ง MPL มักมีการเปลี่ยนแปลงอื่นๆ ในโครโมโซมด้วย ความผิดปกติทางพันธุกรรมเหล่านี้อาจแสดงการเพิ่มขึ้นอย่างผิดปกติของสารพันธุกรรมบางส่วนที่พบได้ตามปกติบนโครโมโซม 1, 6, 7, 8, 19, 21 และ/หรือ X และการสูญเสียสารพันธุกรรมบางส่วนที่พบได้ตามปกติบนโครโมโซม 2, 3, 4, 5, 10, 11, 12, 13, 14, 15, 16, 17 และ/หรือ 22 สารพันธุกรรมที่หายไปในแถบที่ 14 บนแขนยาวของโครโมโซม 13 นั้นไม่เพียงแต่รวมถึง ยีน RP1 เท่านั้น แต่ยังรวม ถึงยีน RCBTB2 , DLEU1และITM2B ด้วย เนื่องจากความหายากและคำจำกัดความที่เพิ่งเกิดขึ้นใหม่ ลักษณะทางโมเลกุลและความสำคัญของความผิดปกติทางพันธุกรรมเหล่านี้จึงยังไม่ได้รับการกำหนดอย่างครบถ้วน[ 56 ] อย่างไรก็ตาม การศึกษาชี้ให้เห็นว่าการสูญเสียยีน RB1, RCBTB2, DLEU1และITM2Bอย่างน้อยหนึ่งยีนแต่โดยเฉพาะอย่างยิ่ง ยีน RP1อาจมีส่วนเกี่ยวข้องในการพัฒนาและ/หรือความก้าวหน้าของ MPL [ 62 ]
การวินิจฉัย
การวินิจฉัย MPL ขึ้นอยู่กับลักษณะทางคลินิกของเนื้องอก ความคล้ายคลึงทางพยาธิวิทยาของเนื้อเยื่อกับ myxoid liposarcoma และที่สำคัญที่สุดคือการไม่มี ยีนฟิวชั่น FUS-DDIT3 sn EWSR1-DDIT3ในเซลล์เนื้องอก[ 62 ] [ 6 ]
การรักษาและการพยากรณ์โรค
แม้ว่าผู้ป่วย MPL จะได้รับการรักษาด้วยการผ่าตัดเพื่อเอาเนื้องอกออก[ 64 ] [ 65 ] [ 6 ] [ 66 ]แต่การทบทวนในปี 2021 พบว่าไม่มีข้อแนะนำที่เป็นเอกฉันท์สำหรับมาตรฐานการดูแล MPL ในส่วนที่เกี่ยวกับการรักษาด้วยรังสีและเคมีบำบัด (ไม่ว่าจะใช้เพียงอย่างเดียวหรือร่วมกับการผ่าตัด) สำหรับการรักษาเนื้องอกเหล่านี้[ 6 ]
พยาธิวิทยาของเนื้องอกไขมันชนิดลิโปซาร์โคมา
 รูปที่ 1 ภาพจุลทรรศน์แสดงการสร้างกระดูกในเนื้องอกไลโปซาร์โคมา
รูปที่ 1 ภาพจุลทรรศน์แสดงการสร้างกระดูกในเนื้องอกไลโปซาร์โคมา รูปที่ 2 ภาพจุลทรรศน์ของเนื้องอกไลโปซาร์โคมาที่เสื่อมสภาพ
รูปที่ 2 ภาพจุลทรรศน์ของเนื้องอกไลโปซาร์โคมาที่เสื่อมสภาพ รูปที่ 3 ภาพจุลทรรศน์ กำลังขยายต่ำ ของเนื้องอกไมซอยด์ไลโปซาร์โคมา
รูปที่ 3 ภาพจุลทรรศน์ กำลังขยายต่ำ ของเนื้องอกไมซอยด์ไลโปซาร์โคมา รูปที่ 4 ภาพจุลทรรศน์ กำลังขยายสูง ของเนื้องอกไมซอยด์ไลโปซาร์โคมา
รูปที่ 4 ภาพจุลทรรศน์ กำลังขยายสูง ของเนื้องอกไมซอยด์ไลโปซาร์โคมา




การถ่ายภาพทางการแพทย์
การตรวจอัลตราซาวนด์ทางการแพทย์และการถ่ายภาพด้วยคลื่นแม่เหล็กไฟฟ้า (MRI) ของลิโปซาร์โคมามีประโยชน์และมักจำเป็นในการพิจารณาขอบเขต การเข้าถึงการผ่าตัด และความสัมพันธ์กับความผิดปกติของอวัยวะที่สังเกตได้ เนื่องจากการตรวจอัลตราซาวนด์มักไม่สามารถแยกแยะลิโปซาร์โคมาออกจากลิโปมาที่ไม่เป็นอันตรายได้ ดังนั้น MRI จึงเป็นวิธีการถ่ายภาพเบื้องต้นที่เลือกใช้เพื่อให้ได้หลักฐานที่เกี่ยวข้องกับการแยกแยะความแตกต่างนี้[ 67 ]
ในมะเร็งไขมันชนิดมิกซอยด์ จะแสดงมวลที่มีความเข้มสัญญาณต่ำและมีจุดที่มีความเข้มสัญญาณสูงในภาพ MRI แบบ T1-weighted มวลจะแสดงความเข้มสัญญาณสูงในภาพแบบ T2-weighted เนื่องจากมีสารเมือก เป็นส่วนใหญ่ (ทำให้มีความเข้มสัญญาณต่ำในภาพ T1) และมีไขมันที่เจริญเต็มที่ในปริมาณเล็กน้อย (ทำให้มีความเข้มสัญญาณสูงในภาพ T1) [ 68 ]มวลมีขอบเขตชัดเจน เป็นพู เป็นหลายช่อง หรือเป็นรูปไข่ โดยไม่มีการแทรกซึมเข้าไปในโครงสร้างโดยรอบ[ 68 ]
![รูปที่ 5 การตรวจอัลตราซาวนด์ของลิโปซาร์โคมาที่มีบริเวณสะท้อนเสียงสูงซึ่งสะท้อนจากเมทริกซ์ไขมันและบริเวณสะท้อนเสียงต่ำซึ่งสะท้อนจากบริเวณที่ไม่ใช่ไขมัน[69]](//upload.wikimedia.org/wikipedia/commons/thumb/a/a9/Scrotal_ultrasonography_of_liposarcoma.jpg/120px-Scrotal_ultrasonography_of_liposarcoma.jpg) รูปที่ 5 การตรวจอัลตราซาวนด์ของลิโปซาร์โคมาที่มีบริเวณสะท้อนเสียงสูงซึ่งสะท้อนจากเมทริกซ์ไขมันและบริเวณสะท้อนเสียงต่ำซึ่งสะท้อนจากบริเวณที่ไม่ใช่ไขมัน[ 69 ]
รูปที่ 5 การตรวจอัลตราซาวนด์ของลิโปซาร์โคมาที่มีบริเวณสะท้อนเสียงสูงซึ่งสะท้อนจากเมทริกซ์ไขมันและบริเวณสะท้อนเสียงต่ำซึ่งสะท้อนจากบริเวณที่ไม่ใช่ไขมัน[ 69 ]![รูปที่ 6 การตรวจอัลตราซาวนด์ของลิโปซาร์โคมาที่เลียนแบบลิโปมา ก้อนเนื้อที่มีความหนาแน่นสูงและมีความสะท้อนเสียงสูงนี้มีลักษณะเหมือนกับลิโปมา[69]](//upload.wikimedia.org/wikipedia/commons/thumb/3/34/Scrotal_ultrasonography_of_liposarcoma_mimicking_a_lipoma.jpg/120px-Scrotal_ultrasonography_of_liposarcoma_mimicking_a_lipoma.jpg) รูปที่ 6 การตรวจอัลตราซาวนด์ของลิโปซาร์โคมาที่เลียนแบบลิโปมา ก้อนเนื้อที่มีความหนาแน่นสูงและสม่ำเสมอนี้มีลักษณะเหมือนกับลิโปมา[ 69 ]
รูปที่ 6 การตรวจอัลตราซาวนด์ของลิโปซาร์โคมาที่เลียนแบบลิโปมา ก้อนเนื้อที่มีความหนาแน่นสูงและสม่ำเสมอนี้มีลักษณะเหมือนกับลิโปมา[ 69 ] ภาพที่ 7 ภาพ MRIของเนื้องอกไขมันชนิดมิกซอยด์เกรดสูง บริเวณรักแร้ด้าน ซ้าย ของชายอายุ 40 ปี ซึ่งเน้นด้วยสีขาว ในภาพตัดขวางของเนื้องอก
ภาพที่ 7 ภาพ MRIของเนื้องอกไขมันชนิดมิกซอยด์เกรดสูง บริเวณรักแร้ด้าน ซ้าย ของชายอายุ 40 ปี ซึ่งเน้นด้วยสีขาว ในภาพตัดขวางของเนื้องอก
![รูปที่ 5 การตรวจอัลตราซาวนด์ของลิโปซาร์โคมาที่มีบริเวณสะท้อนเสียงสูงซึ่งสะท้อนจากเมทริกซ์ไขมันและบริเวณสะท้อนเสียงต่ำซึ่งสะท้อนจากบริเวณที่ไม่ใช่ไขมัน[69]](https://upload.wikimedia.org/wikipedia/commons/thumb/a/a9/Scrotal_ultrasonography_of_liposarcoma.jpg/120px-Scrotal_ultrasonography_of_liposarcoma.jpg)
![รูปที่ 6 การตรวจอัลตราซาวนด์ของลิโปซาร์โคมาที่เลียนแบบลิโปมา ก้อนเนื้อที่มีความหนาแน่นสูงและมีความสะท้อนเสียงสูงนี้มีลักษณะเหมือนกับลิโปมา[69]](https://upload.wikimedia.org/wikipedia/commons/thumb/3/34/Scrotal_ultrasonography_of_liposarcoma_mimicking_a_lipoma.jpg/120px-Scrotal_ultrasonography_of_liposarcoma_mimicking_a_lipoma.jpg)

สังคมและวัฒนธรรม
กรณีที่น่าสนใจ
- แชด บราวน์ (1961–2014) นักเล่นโป๊กเกอร์ เสียชีวิตด้วยโรคมะเร็งไขมัน (liposarcoma)
- ริชาร์ด เฟย์นแมน (1918–1988) นักฟิสิกส์ทฤษฎี เสียชีวิตหลังจากการผ่าตัดเพื่อรักษาโรคดังกล่าว
- ร็อบ ฟอร์ด (1969–2016) อดีตนายกเทศมนตรีและสมาชิกสภาเมืองโทรอนโต เสียชีวิตด้วยโรคมะเร็งไขมันชนิดเพลโอโมฟิก
- โฮกี้ กาจาน (1959–2016) อดีตรันนิ่งแบ็กของทีมนิวออร์ลีนส์ เซนต์สและผู้บรรยายเกมทางวิทยุของทีม เสียชีวิตด้วยโรคมะเร็งไขมัน (liposarcoma)
- ชาร์ลี เดวีส์ (เกิดปี 1986) อดีตนักฟุตบอลของทีมฟิลาเดลเฟีย ยูเนียนในเมเจอร์ลีกซอกเกอร์ ได้รับการวินิจฉัยว่าเป็นมะเร็งไขมันชนิดไลโปซาร์โคมาในปี 2016
- มาร์ค สแตรนด์ (1934–2014) อดีตกวีเอกของสหรัฐอเมริกาและผู้ได้รับรางวัลพูลิตเซอร์ เสียชีวิตด้วยโรคมะเร็งไขมัน (liposarcoma)
ดูเพิ่มเติม
- ลิโปมา
- มิลาเดเมแทนยาใหม่ที่อยู่ระหว่างการวิจัย
- เวนดี้ วอล์ค (Wendy Walk)เป็นองค์กรไม่แสวงหาผลกำไรที่มีพันธกิจในการระดมทุนและสร้างความตระหนักรู้เกี่ยวกับมะเร็งเนื้อเยื่อเกี่ยวพัน รวมถึงมะเร็งไขมัน (liposarcoma)
สรุปเนื้อหา
ข้อมูลสำคัญจากบทความ
ข้อมูลสำคัญเกี่ยวกับ ลิโปซาร์โคมา
ลิโปซาร์โคมาเป็นชนิดย่อยที่พบบ่อยที่สุดของซาร์โคมา เนื้อเยื่ออ่อน คิดเป็นอย่างน้อย 20% ของซาร์โคมาทั้งหมดในผู้ใหญ่ซาร์โคมาเนื้อเยื่ออ่อนเป็นเนื้องอก ที่หายาก มี...
นิรุกติศาสตร์
"เนื้องอกไขมัน" (พหูพจน์ lipomata) ค.ศ. 1830 มาจากภาษาละตินทางการแพทย์ มาจากภาษากรีก lipos "ไขมัน" (คำนาม) มาจากรากศัพท์ PIE *leip- "ติด, เกาะ" ซึ่งใช้ในการสร้างคำที่หมายถึง "ไขมัน" เช่นกัน + -oma
รูปแบบของลิโปซาร์โคมา
โดยทั่วไปแล้วลิโปซาร์โคมาเป็นเนื้องอกขนาดใหญ่ (>10 ซม.) แต่สามารถมีขนาดได้เกือบทุกขนาด ส่วนใหญ่เกิดขึ้นในผู้ใหญ่ โดยมีเพียง 0.
เนื้องอกไขมันชนิดผิดปกติ/มะเร็งไขมันชนิดที่มีการแยกแยะเซลล์ได้ดี
โดยรวมแล้ว เนื้องอกไขมันผิดปกติ (ALTs) และลิโปซาร์โคมาที่มีการแยกแยะเซลล์ได้ดี (WDLs) คิดเป็น 40–45% ของลิโปซาร์โคมาทั้งหมด [ 11 ] เนื้องอก เหล่านี้แทบจะไม่แพร่ กระจายไป ยังส่วนอื่นของร่างกาย เลย ดังนั้นจึงถือว่าเป็นเนื้องอก ที่ไม่ร้ายแรง หรือ...