โรคฮันติงตัน
| โรคฮันติงตัน | |
|---|---|
| ชื่ออื่นๆ | โรคฮันติงตัน |
 | |
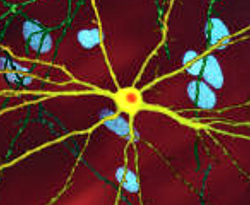
| ภาพถ่ายจุลทรรศน์ที่ได้รับการแก้ไขของเซลล์ประสาทหนามขนาดกลาง (สีเหลือง) ที่มีสิ่งเจือปน (สีส้ม) ซึ่งเกิดขึ้นเป็นส่วนหนึ่งของกระบวนการเกิดโรค (ความกว้างของภาพ 360 ไมโครเมตร ) | |
| ความเชี่ยวชาญ | ประสาทวิทยา , พันธุศาสตร์การแพทย์ |
| อาการ | ปัญหาเกี่ยวกับทักษะการเคลื่อนไหว รวมถึงการประสานงานและการเดิน อารมณ์ และความสามารถทางจิต[ 1 ] [ 2 ] |
| ภาวะแทรกซ้อน | โรคปอดบวมโรคหัวใจการบาดเจ็บทางร่างกายจากการหกล้มการฆ่าตัวตาย[ 3 ] |
| เริ่มต้นตามปกติ | อายุ 30–50 ปี[ 4 ] |
| ระยะเวลา | ระยะยาว[ 4 ] |
| สาเหตุ | พันธุกรรม (ที่สืบทอดมาหรือการกลายพันธุ์ใหม่) [ 4 ] |
| วิธีการวินิจฉัย | การทดสอบทางพันธุกรรม[ 5 ] |
| การวินิจฉัยแยกโรค | โรค Sydenham's chorea , โรค chorea ทางพันธุกรรมที่ไม่ร้ายแรง , โรค ลูปัส , กลุ่มอาการ paraneoplastic , โรค Wilson [ 6 ] |
| การรักษา | การดูแลสนับสนุน[ 2 ] |
| ยา | เตตระเบนาซีน[ 3 ] |
| การพยากรณ์โรค | 15–20 ปีนับจากเริ่มมีอาการ ซึ่งมักจะถึงแก่ความตาย[ 4 ] |
| ความถี่ | 4–15 ใน 100,000 (เชื้อสายยุโรป) [ 1 ] |
| ตั้งชื่อตาม | จอร์จ ฮันติงตัน |
โรคฮันติงตัน ( HD ) หรือที่รู้จักกันในชื่อโรคฮันติงตันโคเรีย เป็นโรคทางระบบประสาทเสื่อมที่ ร้ายแรงถึงแก่ชีวิต [ 7 ]ซึ่งมัก ถ่ายทอด ทางพันธุกรรม[ 8 ]โดยทั่วไปจะแสดงอาการเป็นกลุ่มอาการทางจิตเวช การรับรู้ และการเคลื่อนไหวที่ค่อยๆ รุนแรงขึ้น[ 9 ]อาการแรกเริ่มมักเป็นปัญหาเล็กน้อยเกี่ยวกับอารมณ์หรือความสามารถทางจิต/จิตเวช ซึ่งเกิดขึ้นก่อนอาการทางการเคลื่อนไหวในหลายๆ คน[ 10 ] [ 1 ] ในที่สุด อาการทางกายภาพที่ชัดเจน รวมถึงการขาดการประสานงาน โดยทั่วไป และการเดิน ที่ไม่มั่นคง ก็จะตามมา[ 2 ]เมื่อเวลาผ่านไปบริเวณฐานสมอง จะ ค่อยๆถูกทำลาย[ 11 ]
โรคนี้มีลักษณะเด่นคือความผิดปกติของการเคลื่อนไหว แบบ ไฮเปอร์ไค เนติก ที่เรียกว่าโคเรีย [ 12 ] [ 13 ] โคเรียมักแสดงออกเป็นการเคลื่อนไหวของร่างกายที่ไม่ประสานกัน ไม่ได้ตั้งใจ และ "คล้ายการเต้นรำ" ซึ่งจะเห็นได้ชัดเจนมากขึ้นเมื่อโรคดำเนินไป[ 1 ]ความสามารถทางกายภาพจะค่อยๆ แย่ลงจนกระทั่งการเคลื่อนไหวที่ประสานกันกลายเป็นเรื่องยากและบุคคลนั้นไม่สามารถพูดได้[ 1 ] [ 2 ]ความสามารถทางจิตใจโดยทั่วไปจะลดลงจนเกิดภาวะสมองเสื่อม ภาวะซึมเศร้า เฉื่อยชา และหุนหันพลันแล่นในบางครั้ง[ 10 ] [ 14 ] [ 3 ]อาการเฉพาะจะแตกต่างกันไปบ้างในแต่ละกรณี[ 1 ]อาการสามารถเริ่มต้นได้ทุกวัย แต่มักจะพบครั้งแรกเมื่ออายุประมาณ 40 ปี[ 14 ] [ 10 ] [ 3 ] [ 4 ]โรคอาจพัฒนาเร็วขึ้นในแต่ละรุ่นที่สืบต่อกันมา[ 1 ]ประมาณร้อยละ 8 ของผู้ป่วยเริ่มมีอาการก่อนอายุ 20 ปี และเรียกว่าHD ในเด็กซึ่งมักแสดงอาการเคลื่อนไหวช้าแบบโรคพาร์กินสันมากกว่าอาการชักกระตุก[ 3 ]
โดยทั่วไปแล้ว HD มักถ่ายทอดทางพันธุกรรมจากพ่อแม่ที่เป็นโรคซึ่งมียีนกลายพันธุ์ในยีน huntingtin ( HTT ) ซึ่งเป็นยีนที่สร้าง โปรตีนhuntingtin [ 4 ]อย่างไรก็ตาม มากถึง 10% ของกรณีเกิดจากการกลายพันธุ์ใหม่[ 1 ]การขยายตัวของCAG ซ้ำ (ที่รู้จักกันในชื่อการขยายตัวของไตรนิวคลีโอไทด์ซ้ำ ) ในยีนที่สร้างโปรตีน huntingtin ส่งผลให้เกิดโปรตีนกลายพันธุ์ที่ผิดปกติ (mHTT) ซึ่งค่อยๆ ทำลายเซลล์สมองผ่านกลไกต่างๆ ที่เป็นไปได้[ 8 ] [ 15 ]โปรตีนกลายพันธุ์นี้เป็นลักษณะเด่นดังนั้นการมีพ่อหรือแม่ที่เป็นพาหะของลักษณะนี้ก็เพียงพอที่จะกระตุ้นให้เกิดโรคในลูกได้ การวินิจฉัยทำได้โดยการตรวจทางพันธุกรรมซึ่งสามารถทำได้ทุกเมื่อ โดยไม่คำนึงว่าจะมีอาการหรือไม่[ 5 ]ข้อเท็จจริงนี้ก่อให้เกิดการถกเถียงทางจริยธรรมหลายประการ ได้แก่ อายุที่บุคคลถือว่ามีวุฒิภาวะเพียงพอที่จะเลือกการตรวจ พ่อแม่มีสิทธิที่จะให้ลูกได้รับการตรวจหรือไม่ และการจัดการความลับและการเปิดเผยผลการตรวจ[ 2 ]
ยังไม่มีวิธีรักษาโรค HD ที่ได้ผล และจำเป็นต้องได้รับการดูแลตลอดเวลาในระยะหลัง[ 2 ]การรักษาสามารถบรรเทาอาการบางอย่างและอาจช่วยปรับปรุงคุณภาพชีวิตได้[ 3 ]หลักฐานที่ดีที่สุดสำหรับการรักษาปัญหาการเคลื่อนไหวคือการใช้เททราเบนาซีน [ 3 ] โรค HD ส่งผลกระทบต่อประชากรเชื้อสายยุโรปประมาณ 4 ถึง 15 คนต่อ 100,000 คน[ 1 ] [ 3 ]พบได้น้อยในชาวฟินแลนด์และชาวญี่ปุ่น ในขณะที่อุบัติการณ์ในแอฟริกายังไม่ทราบ[ 3 ]โรคนี้ส่งผลกระทบต่อทั้งชายและหญิงเท่าๆ กัน[ 3 ]ภาวะแทรกซ้อน เช่นปอดบวมโรคหัวใจและการบาดเจ็บทางร่างกายจากการหกล้ม ทำให้อายุขัยลดลง โรคปอดบวมจากการสำลักที่ร้ายแรงมักถูกกล่าวถึงว่าเป็นสาเหตุการเสียชีวิตขั้นสุดท้ายของผู้ป่วย[ 16 ] [ 14 ] [ 3 ]การฆ่าตัวตายเป็นสาเหตุการเสียชีวิตประมาณ 9% ของผู้ป่วย[ 3 ]โดยทั่วไปผู้ป่วยจะเสียชีวิตภายใน 15-20 ปีนับจากวันที่ตรวจพบโรคครั้งแรก[ 4 ]
คำอธิบายที่เก่าแก่ที่สุดเกี่ยวกับโรคนี้ได้รับการบันทึกไว้ในปี พ.ศ. 2484 โดยแพทย์ชาวอเมริกัน Charles Oscar Waters [ 17 ] แพทย์ชาวอเมริกัน George Huntington ได้อธิบายสภาพของโรคนี้โดยละเอียดเพิ่มเติมในปี พ.ศ. 2415 [ 17 ] พื้นฐานทางพันธุกรรมถูกค้นพบในปี พ.ศ. 2536 โดยความร่วมมือระหว่างประเทศที่นำโดยมูลนิธิโรคทางพันธุกรรม [ 18 ] [ 19 ]องค์กรวิจัยและสนับสนุนเริ่มก่อตั้งขึ้นในช่วงปลายทศวรรษพ.ศ. 2503เพื่อเพิ่มความตระหนักรู้ของสาธารณชน ให้การสนับสนุนผู้ป่วยและครอบครัว และส่งเสริมการวิจัย[ 19 ] [ 20 ]ทิศทางการวิจัยรวมถึงการกำหนดกลไกที่แน่นอนของโรค การปรับปรุงแบบจำลองสัตว์เพื่อช่วยในการวิจัย การทดสอบยาและการส่งมอบ ยา เพื่อรักษาอาการหรือชะลอการลุกลามของโรค และการศึกษาขั้นตอนต่างๆ เช่นการบำบัดด้วยสเต็มเซลล์โดยมีเป้าหมายเพื่อทดแทนเซลล์ประสาทที่เสียหายหรือสูญเสียไป[ 18 ]
อาการและสัญญาณ
อาการและสัญญาณของโรคฮันติงตันมักจะสังเกตเห็นได้ชัดเจนในช่วงอายุ 30 ถึง 50 ปี แต่สามารถเริ่มได้ทุกช่วงอายุ[ 4 ]และแสดงออกมาในรูปแบบของอาการสามอย่าง ได้แก่ อาการทางมอเตอร์ อาการทางสติปัญญา และอาการทางจิตเวช[ 21 ]หากเกิดขึ้นในระยะเริ่มต้น จะเรียกว่าโรคฮันติงตันในวัยเด็ก[ 22 ]ใน 50% ของกรณี อาการทางจิตเวชจะปรากฏขึ้นก่อน[ 21 ]ซึ่งอาจเกิดขึ้นก่อนอาการทางมอเตอร์หลายปี[ 23 ] [ 24 ]ความคืบหน้าของโรคมักจะถูกอธิบายในระยะเริ่มต้น ระยะกลาง และระยะท้าย โดยมีระยะนำ ก่อน [ 2 ]ในระยะเริ่มต้น จะมีการเปลี่ยนแปลงบุคลิกภาพเล็กน้อย ปัญหาด้านการรับรู้และทักษะทางกายภาพความหงุดหงิดและอารมณ์แปรปรวน ซึ่งทั้งหมดนี้อาจไม่เป็นที่สังเกตเห็น[ 25 ] [ 26 ]เกือบทุกคนที่เป็นโรค HD จะแสดงอาการทางกายภาพที่คล้ายคลึงกันในที่สุด แต่การเริ่มต้น ความก้าวหน้า และขอบเขตของอาการทางด้านการรับรู้และพฤติกรรมจะแตกต่างกันอย่างมากในแต่ละบุคคล[ 27 ] [ 28 ]
อาการทางกายภาพเริ่มต้นที่เด่นชัดที่สุดคือการเคลื่อนไหวที่กระตุก สุ่ม และควบคุมไม่ได้ เรียกว่าโคเรีย [ 12 ] หลายคนไม่รู้ตัวถึงการเคลื่อนไหวโดยไม่ตั้งใจ หรือถูกขัดขวางโดยการเคลื่อนไหวเหล่านั้น[ 1 ]โคเรียอาจแสดงออกมาในระยะเริ่มต้นในรูปแบบของความกระสับกระส่ายทั่วไป การเคลื่อนไหวเล็กๆ น้อยๆ ที่เริ่มต้นโดยไม่ตั้งใจหรือไม่สมบูรณ์ การขาดการประสานงาน หรือการเคลื่อนไหวของดวงตาแบบ saccadic ที่ช้าลง[ 29 ]ความผิดปกติทางการเคลื่อนไหวเล็กน้อยเหล่านี้มักจะเกิดขึ้นก่อนสัญญาณที่ชัดเจนมากขึ้นของการทำงานผิดปกติของการเคลื่อนไหวอย่างน้อยสามปี[ 30 ]อาการที่ปรากฏอย่างชัดเจน เช่น ความแข็งเกร็ง การเคลื่อนไหวบิดตัว หรือท่าทางที่ผิดปกติจะปรากฏขึ้นเมื่อความผิดปกติดำเนินไป[ 29 ]สิ่งเหล่านี้เป็นสัญญาณว่าระบบในสมองที่รับผิดชอบการเคลื่อนไหวได้รับผลกระทบ[ 31 ] การทำงาน ของจิตและกล้ามเนื้อจะบกพร่องมากขึ้นเรื่อยๆ จนกระทั่งการกระทำใดๆ ที่ต้องใช้การควบคุมกล้ามเนื้อได้รับผลกระทบ เมื่อการควบคุมกล้ามเนื้อได้รับผลกระทบ เช่น ความแข็งเกร็งหรือกล้ามเนื้อหดเกร็ง จะเรียกว่าดิสโทเนีย โรคดิสโท เนียเป็นความผิดปกติของการเคลื่อนไหวแบบไฮเปอร์ไคเนติกทางระบบประสาท ซึ่งส่งผลให้เกิดการบิดตัวหรือการเคลื่อนไหวซ้ำๆ ที่อาจคล้ายกับอาการสั่น ผลกระทบที่พบบ่อย ได้แก่ ความไม่เสถียรทางกายภาพ การแสดงออกทางสีหน้าที่ผิดปกติ และความยากลำบากในการเคี้ยวกลืนและพูด[ 29 ]การนอนหลับไม่ปกติ[ 32 ]และการลดน้ำหนักก็เป็นอาการที่เกี่ยวข้องเช่นกัน และความยากลำบากในการรับประทานอาหารอาจทำให้เกิดการลดน้ำหนักและภาวะทุพโภชนาการ[ 33 ] [ 34 ] [ 35 ]โรค HD ในวัยเด็กมักจะดำเนินไปในอัตราที่เร็วกว่าพร้อมกับการลดลงของความรู้ความเข้าใจที่มากขึ้น และอาการชักแบบโคเรียจะแสดงออกมาเพียงช่วงสั้นๆ หรืออาจไม่แสดงเลย รูปแบบเวสต์ฟาลของการเคลื่อนไหวช้า ความแข็งเกร็ง และอาการสั่นเป็นลักษณะทั่วไปของโรค HD ในวัยเด็ก เช่นเดียวกับอาการชัก[ 29 ] [ 33 ]
ความสามารถทางปัญญาจะเสื่อมถอยลงเรื่อยๆ และมีแนวโน้มที่จะเสื่อมถอยลงจนกลายเป็นภาวะสมองเสื่อม [ 3 ] โดย เฉพาะอย่างยิ่ง หน้าที่การบริหารจัดการจะได้รับผลกระทบซึ่งรวมถึงการวางแผน ความยืดหยุ่นทางปัญญาการคิดเชิงนามธรรม การเรียนรู้กฎเกณฑ์ การเริ่มต้นการกระทำที่เหมาะสม และการยับยั้งการกระทำที่ไม่เหมาะสม ความบกพร่องทางปัญญาต่างๆ ได้แก่ ความยากลำบากในการจดจ่อกับงาน การขาดความยืดหยุ่น การขาดแรงกระตุ้น การขาดความตระหนักรู้เกี่ยวกับพฤติกรรมและความสามารถของตนเอง และความยากลำบากในการเรียนรู้หรือประมวลผลข้อมูลใหม่ เมื่อโรคดำเนินไป ความบกพร่อง ทางความจำมักจะปรากฏขึ้น ความบกพร่องที่รายงานมีตั้งแต่ความบกพร่องของความจำระยะสั้นไป จนถึงความยากลำบาก ของความจำระยะยาวรวมถึงความบกพร่องใน ความจำ แบบเหตุการณ์ (ความจำเกี่ยวกับชีวิตของตนเอง) ความจำ แบบขั้นตอน (ความจำเกี่ยวกับร่างกายว่าต้องทำอย่างไรจึงจะทำกิจกรรมได้) และความจำใช้งาน[ 31 ]
อาการทางจิตเวชที่รายงาน ได้แก่ ความวิตกกังวลภาวะซึมเศร้าการแสดงอารมณ์ที่ลดลง การ เห็นแก่ตัว ความก้าวร้าวพฤติกรรมย้ำคิด ย้ำทำ และอาการ ประสาทหลอน และหลงผิด [ 36 ] ความผิดปกติทางจิตเวชทั่วไปอื่นๆ อาจรวมถึงโรคย้ำคิดย้ำทำ โรคอารมณ์แปรปรวนโรคนอนไม่หลับและโรคอารมณ์สองขั้วนอกจากนี้ยังพบความยากลำบากในการรับรู้การแสดงออกเชิงลบของผู้อื่นด้วย[ 31 ]ความชุกของอาการเหล่านี้แตกต่างกันอย่างมากในแต่ละการศึกษา โดยมีอัตราความชุกตลอดชีวิตของความผิดปกติทางจิตเวช โดยประมาณ อยู่ระหว่าง 33 ถึง 76% [ 36 ]สำหรับผู้ป่วยและครอบครัวจำนวนมาก อาการเหล่านี้เป็นหนึ่งในแง่มุมที่สร้างความทุกข์ทรมานมากที่สุดของโรค มักส่งผลกระทบต่อการทำงานในชีวิตประจำวันและเป็นเหตุผลสำหรับการเข้ารับการรักษาในสถาบัน [ 36 ] การเปลี่ยนแปลงพฤติกรรมในระยะเริ่มต้นของ HD ส่งผลให้มีความเสี่ยงต่อการฆ่าตัวตายเพิ่มขึ้น[ 12 ]บ่อยครั้งที่ผู้ป่วยมีความตระหนักรู้เกี่ยวกับอาการชักกระตุก ความบกพร่องทางด้านการรับรู้และอารมณ์ลดลง[ 37 ]
พันธุศาสตร์
โรคฮันติงตันเป็นความผิดปกติของการทำซ้ำของไตรนิวคลีโอไทด์ที่เกิดจากการขยายตัวของการทำซ้ำ ของไตรนิ วคลีโอไท ด์ ในเอ็กซอนแรกของ ยีนฮันติงติน ( HTT ) ซึ่งเข้ารหัสโปรตีนฮันติงติน (HTT) [ 29 ] HTTอาจถูกเรียกว่ายีน HD หรือในอดีตเรียกว่าIT15 (interesting transcript 15) ตั้งอยู่บนแขนสั้นของโครโมโซม 4 [ 29 ]ที่ 4p16.3 บริเวณการทำซ้ำของไตรนิวคลีโอไทด์ของHTTส่วนใหญ่ประกอบด้วย CAG ซึ่งเข้ารหัสกรดอะมิโนกลูตามีนดังนั้นโปรตีนที่ได้จึงมีส่วนของโพลีกลูตามีน (polyQ tract) [ 38 ]จำนวนการทำซ้ำมีความยาวแตกต่างกันไปในแต่ละบุคคลและอาจเปลี่ยนแปลงความยาวระหว่างรุ่น เมื่อความยาวของส่วนที่ทำซ้ำนี้เกินเกณฑ์ที่กำหนด จะทำให้เกิดโปรตีนฮันติงตินกลายพันธุ์ (mHTT) ซึ่ง mHTT มีผลเสียต่อการทำงานของเซลล์และนำไปสู่โรค การกลายพันธุ์ของโรคฮันติงตันเป็นลักษณะเด่นทางพันธุกรรมและแสดงอาการ เกือบสมบูรณ์ อัลลีล HTT ที่ กลายพันธุ์เพียงตัวเดียวจากพ่อหรือแม่ก็เพียงพอที่จะทำให้เกิดโรคได้[ 29 ]เนื่องจากอัตราการกลายพันธุ์สูงกว่าในเซลล์อสุจิ เพศชายจึงมีแนวโน้มที่จะถ่ายทอดอัลลีล HTT ที่ขยายตัวไปยังลูกหลานมากกว่า[ 39 ]

| การนับซ้ำ | การจำแนกประเภท | สถานะของโรค | ความเสี่ยงต่อลูกหลาน |
|---|---|---|---|
| <27 | ปกติ | จะไม่ได้รับผลกระทบ | ไม่มี |
| 27–35 | ระดับกลาง | จะไม่ได้รับผลกระทบ | สูงขึ้น แต่ต่ำกว่า 50% |
| 36–39 | การแทรกซึมที่ลดลง | อาจได้รับผลกระทบหรือไม่ก็ได้ | 50% |
| 40+ | การแทรกซึมเต็มที่ | จะได้รับผลกระทบ | 50% |
โดยทั่วไป ผู้คนจะมีจำนวนการทำซ้ำในบริเวณโพลีคิว ( polyQ) น้อยกว่า 36 ครั้ง ซึ่งส่งผลให้มีการผลิตและ การแปลตำแหน่ง ไซโตพลาสมิกของฮันติงติน ตามปกติ [ 29 ] อย่างไรก็ตาม การทำซ้ำ 36 ครั้งขึ้นไปจะส่งผลให้เกิดการผลิตฮันติงตินกลายพันธุ์ (mHTT) [ 29 ]ซึ่งทำให้เกิดการเสื่อมของเซลล์ประสาทหนามขนาดกลางบริเวณต่างๆ ของสมองมีปริมาณและการพึ่งพาเซลล์ประสาทประเภทนี้แตกต่างกัน และได้รับผลกระทบตามนั้น[ 29 ]โดยทั่วไป จำนวนการทำซ้ำของ CAG เกี่ยวข้องกับว่ากระบวนการนี้ได้รับผลกระทบมากน้อยเพียงใด และคิดเป็นประมาณ 60% ของความแปรปรวนของอายุที่เริ่มมีอาการ ความแปรปรวนที่เหลือเกิดจากสิ่งแวดล้อมและยีนอื่นๆ ที่ปรับเปลี่ยนกลไกของ HD [ 29 ]การทำซ้ำประมาณ 36 ถึง 39 ครั้งส่งผลให้เกิดโรคในรูปแบบที่มีการแทรกซึมลดลง โดยมีอาการเริ่มช้ากว่ามากและดำเนินไปช้ากว่า ในบางกรณี อาการเริ่มช้ามากจนไม่เคยสังเกตเห็นเลย[ 29 ]ด้วยจำนวนการทำซ้ำที่มาก (มากกว่า 60) โรค HD สามารถเริ่มแสดงอาการได้ก่อนอายุ 20 ปี ซึ่งเรียกว่า HD ในวัยเยาว์ โดยทั่วไปแล้ว HD ในวัยเยาว์จะเป็นแบบ Westphal ซึ่งมีลักษณะเฉพาะคือการเคลื่อนไหวช้า แข็งเกร็ง และสั่น ซึ่งคิดเป็นประมาณ 7% ของผู้ที่เป็นพาหะของ HD [ 40 ] [ 41 ]
มรดก

โรคฮันติงตันมี การถ่ายทอดทางพันธุกรรม แบบออโตโซมัลโดมิแนนต์หมายความว่าผู้ป่วยมักจะได้รับยีนหนึ่งชุดที่มีการทำซ้ำของไตรนิวคลีโอไทด์ที่ขยายตัว ( อัลลีล กลายพันธุ์ ) จากพ่อแม่ที่เป็นโรค[ 29 ]เนื่องจากความสามารถในการแสดงออกของยีนกลายพันธุ์นั้นสูงมาก ผู้ที่มียีนกลายพันธุ์จึงจะเป็นโรค ในรูปแบบการถ่ายทอดทางพันธุกรรมแบบนี้ ลูกหลานของผู้ป่วยแต่ละคนจะมีโอกาส 50% ที่จะได้รับอัลลีลกลายพันธุ์และเป็นโรค (ดูรูป) ความน่าจะเป็นนี้ไม่ขึ้นกับเพศ เนื่องจากยีนฮันติงตินไม่ได้อยู่บนโครโมโซม X หรือ Y [ 42 ] [ 43 ]
การทำซ้ำของไตรนิวคลีโอไทด์ CAGที่มีจำนวนมากกว่า 28 ครั้งนั้นไม่เสถียรในระหว่างการจำลองแบบและความไม่เสถียรนี้จะเพิ่มขึ้นตามจำนวนการทำซ้ำที่มีอยู่[ 29 ]ซึ่งมักจะนำไปสู่การขยายตัวใหม่เมื่อรุ่นต่อๆ ไป ( การกลายพันธุ์แบบไดนามิก ) แทนที่จะสร้างสำเนาที่แน่นอนของการทำซ้ำไตรนิวคลีโอไทด์[ 29 ]สิ่งนี้ทำให้จำนวนการทำซ้ำเปลี่ยนแปลงไปในรุ่นต่อๆ ไป เช่น พ่อแม่ที่ไม่ได้รับผลกระทบที่มีจำนวนการทำซ้ำ "ปานกลาง" (28–35) หรือ "การแทรกซึมที่ลดลง" (36–40) อาจส่งต่อสำเนาของยีนที่มีจำนวนการทำซ้ำเพิ่มขึ้นซึ่งทำให้เกิด HD ที่มีการแทรกซึมอย่างสมบูรณ์[ 29 ]อายุที่เริ่มมีอาการเร็วขึ้นและความรุนแรงของโรคที่มากขึ้นในรุ่นต่อๆ ไปเนื่องจากการเพิ่มขึ้นของจำนวนการทำซ้ำเรียกว่าการคาดการณ์ ทางพันธุกรรม [ 1 ]ความไม่เสถียรจะมากกว่าใน กระบวนการ สร้างสเปิร์มมากกว่ากระบวนการสร้างไข่[ 29 ]อัลลีลที่ถ่ายทอดทางมารดามักจะมีความยาวซ้ำที่คล้ายคลึงกัน ในขณะที่อัลลีลที่ถ่ายทอดทางบิดามีโอกาสสูงที่จะมีความยาวเพิ่มขึ้น[ 29 ] [ 44 ]โรคฮันติงตันเกิดจากการกลายพันธุ์ใหม่ ได้น้อยมาก โดยที่ทั้งพ่อและแม่ไม่มีการทำซ้ำ CAG มากกว่า 36 ครั้ง[ 45 ]
ในสถานการณ์ที่หายากซึ่งทั้งพ่อและแม่มียีน HD ที่ขยายตัว ความเสี่ยงจะเพิ่มขึ้นเป็น 75% และเมื่อพ่อหรือแม่คนใดคนหนึ่งมีสำเนาที่ขยายตัวสองชุด ความเสี่ยงจะอยู่ที่ 100% บุคคลที่มียีนทั้งสองได้รับผลกระทบนั้นหายาก ในช่วงเวลาหนึ่ง HD ถูกคิดว่าเป็นโรคเดียวที่การมียีนกลายพันธุ์ตัวที่สองไม่ส่งผลต่ออาการและการดำเนินของโรค[ 46 ]แต่ต่อมาพบว่าสามารถส่งผลต่อฟีโนไทป์และอัตราการดำเนินของโรคได้[ 29 ] [ 47 ]
กลไก
โปรตีนฮันติงติน (HTT) มีปฏิสัมพันธ์กับโปรตีนอื่นๆ มากกว่า 100 ชนิด และดูเหมือนจะมีหน้าที่หลายอย่าง[ 48 ]พฤติกรรมของโปรตีนกลายพันธุ์ (mHTT) ยังไม่เป็นที่เข้าใจอย่างสมบูรณ์ แต่เป็นพิษต่อเซลล์บางชนิด โดยเฉพาะเซลล์สมองความเสียหายในระยะเริ่มต้นจะเห็นได้ชัดที่สุดในปมประสาทฐานใต้เปลือกสมอง โดยเริ่มแรกใน สไตร อาตัมแต่เมื่อโรคดำเนินไป บริเวณอื่นๆ ของสมองก็ได้รับผลกระทบเช่นกัน รวมถึงบริเวณของเปลือกสมองอาการในระยะเริ่มต้นเกิดจากหน้าที่ของสไตรอาตัมและการเชื่อมต่อกับเปลือกสมอง ได้แก่ การควบคุมการเคลื่อนไหว อารมณ์ และการทำงานของระบบการรับรู้ระดับสูง[ 29 ]การเมทิลเลชั่นของ DNAก็ดูเหมือนจะเปลี่ยนแปลงไปใน HD ด้วย[ 49 ]
ในปี 2025 นักวิทยาศาสตร์จากมหาวิทยาลัยฮาร์วาร์ดและ MIT ได้ตีพิมพ์งานวิจัยที่ตรวจสอบกลไกเบื้องหลังการเกิดอาการ พวกเขาพบว่าความยาวของลำดับไตรนิวคลีโอไทด์ที่ซ้ำกันจะเพิ่มขึ้นตามอายุเนื่องจากข้อผิดพลาดที่สะสมใน กระบวนการ ซ่อมแซม DNA ที่ไม่ตรงกันหลังจากการถอดรหัสและสิ่งนี้จะกลายเป็นพิษเมื่อลำดับขยายเกิน 150 ครั้ง[ 50 ]
หน้าที่ของฮันติงติน
HTT ถูกแสดงออกในเซลล์ทั้งหมด โดยมีความเข้มข้นสูงสุดในสมองและอัณฑะและมีปริมาณปานกลางในตับหัวใจและปอดแม้ว่าขอบเขตการทำงานทั้งหมดของมันจะยังไม่เป็นที่เข้าใจ แต่ HTT มีปฏิสัมพันธ์กับโปรตีนที่เกี่ยวข้องกับการถอดรหัสการส่งสัญญาณของเซลล์และการขนส่งภายในเซลล์[ 51 ] การศึกษาในสัตว์ที่ได้รับการดัดแปลงพันธุกรรมแสดงให้เห็นว่า HTT ชนิดปกติมีความสำคัญต่อการพัฒนาของตัวอ่อน เนื่องจากการขาดหายไปอย่างสมบูรณ์ของยีนนี้ส่งผลให้ตัวอ่อนตาย[ 52 ] HTT ยังมีบทบาทในการปกป้องเซลล์ประสาทที่เจริญเต็มที่: มันควบคุมการผลิตปัจจัยบำรุงเซลล์ประสาทที่ได้จากสมองสนับสนุนการขนส่งถุงเวสิเคิลของไซแนปส์และการส่งสัญญาณไซแนปส์ควบคุมการถอดรหัสยีนของเซลล์ประสาท และป้องกันการตายของเซลล์ตามโปรแกรมโดยการยับยั้ง เอนไซม์ อะพอพโท ซิส เช่นแคสเปส[ 52 ]ตรงกันข้ามกับบทบาทในการป้องกันเหล่านี้ รูปแบบกลายพันธุ์ของโปรตีน (mHTT) จะรบกวนระบบยูบิควิติน-โปรตีเอโซมซึ่งส่งเสริมการกระตุ้นแคสเปส ส่งผลให้เกิดการเสื่อมของระบบประสาท การเพิ่มการแสดงออกของ HTT สามารถส่งเสริม การอยู่รอด ของเซลล์สมองและบรรเทาผลกระทบของ mHTT ในขณะที่การลดการแสดงออก ของ HTT สามารถทำให้ผลกระทบเหล่านั้นรุนแรงขึ้นได้ [ 52 ] ดังนั้น จึงเชื่อว่าโรคนี้ไม่ได้เกิดจาก การแสดงออกของ HTT ที่ไม่เพียงพอ แต่เกิดจากการทำงานที่เป็นพิษของ mHTT ในร่างกาย[ 29 ]โปรตีนกลายพันธุ์นี้แสดงออกทั่วร่างกายและเกี่ยวข้องกับความผิดปกติในเนื้อเยื่อส่วนปลายที่อยู่นอกสมอง ซึ่งอาจรวมถึงกล้ามเนื้อลีบหัวใจล้มเหลวภาวะทนต่อกลูโคสบกพร่องน้ำหนักลดโรคกระดูก พรุน และอัณฑะลีบ[ 53 ]
การเปลี่ยนแปลงระดับเซลล์
การกระทำที่เป็นพิษของ mHTT อาจแสดงออกและก่อให้เกิดพยาธิสภาพของ HD ผ่านการเปลี่ยนแปลงของเซลล์หลายอย่าง[ 54 ] [ 55 ]ในรูปแบบกลายพันธุ์ (โพลีกลูตามีนขยายตัว) โปรตีนมีแนวโน้มที่จะแตกตัวได้ง่ายขึ้น ทำให้เกิดชิ้นส่วนที่สั้นกว่าซึ่งมีการขยายตัวของโพลีกลูตามีน[ 54 ] ชิ้นส่วนโปรตีนเหล่านี้มีแนวโน้มที่จะเกิดการพับตัวผิดปกติและการรวมตัวกัน ทำให้เกิดการรวมตัวเป็นเส้นใยซึ่งสายเบต้าของโพลีกลูตามีนที่ไม่ใช่ของเดิมจากโปรตีนหลายตัวจะเชื่อมต่อกันด้วยพันธะไฮโดรเจน[ 15 ] การรวมตัวเหล่านี้มี สถาปัตยกรรมอะไมลอยด์แบบครอส-เบต้าพื้นฐานเดียวกันกับ ที่พบใน โรคการสะสมโปรตีน อื่น ๆ[ 56 ]เมื่อเวลาผ่านไป การรวมตัวจะสะสมตัวเพื่อสร้างร่างกายรวมภายในเซลล์ ซึ่งในที่สุดจะรบกวนการทำงานของเซลล์ประสาท[ 15 ] [ 54 ]พบร่างกายรวมทั้งใน นิวเคลียส และไซโตพลาสซึมของเซลล์[ 54 ]การรวมตัวของสารในเซลล์ของสมองเป็นหนึ่งในการเปลี่ยนแปลงทางพยาธิวิทยาในระยะแรก และการทดลองบางอย่างพบว่าสารเหล่านี้อาจเป็นพิษต่อเซลล์ แต่การทดลองอื่นๆ แสดงให้เห็นว่าสารเหล่านี้อาจก่อตัวขึ้นเป็นส่วนหนึ่งของกลไกการป้องกันของร่างกายและช่วยปกป้องเซลล์[ 54 ]
มีการระบุเส้นทางหลายเส้นทางที่ mHTT อาจทำให้เซลล์ตาย ซึ่งรวมถึงผลกระทบต่อโปรตีนชาเปอโรนซึ่งช่วยพับโปรตีนและกำจัดโปรตีนที่พับผิดรูป ปฏิสัมพันธ์กับแคสเปสซึ่งมีบทบาทในกระบวนการกำจัดเซลล์ผลกระทบที่เป็นพิษของกลูตามีนต่อเซลล์ประสาทการบกพร่องของการผลิตพลังงานภายในเซลล์ และผลกระทบต่อการแสดงออกของยีน[ 15 ] [ 57 ]
พบว่าโปรตีนฮันติงตินกลายพันธุ์มีบทบาทสำคัญในการทำงานผิดปกติของไมโทคอนเด รี ย[ 51 ] การทำงานบกพร่องของการขนส่งอิเล็กตรอนของไมโทคอนเดรียอาจส่งผลให้เกิดความเครียดออกซิเดชันและการปล่อยอนุมูลอิสระออกซิเจน ในระดับที่สูงขึ้น [ 58 ]
กลูตามีนเป็นที่ทราบกันดีว่ามีฤทธิ์เป็นพิษต่อเซลล์ประสาทเมื่อมีปริมาณมาก ซึ่งอาจก่อให้เกิดความเสียหายต่อโครงสร้างเซลล์จำนวนมาก แม้ว่าจะไม่พบกลูตามีนในปริมาณมากเกินไปในโรค HD แต่การโต้ตอบของโปรตีนฮันติงตินที่เปลี่ยนแปลงไปกับโปรตีนจำนวนมากในเซลล์ประสาททำให้มีความเปราะบางต่อกลูตามีนมากขึ้น ความเปราะบางที่เพิ่มขึ้นนี้เชื่อว่าส่งผลให้เกิดผลกระทบที่เป็นพิษต่อเซลล์ประสาทจากระดับกลูตามีนปกติ[ 15 ]
การขยายตัวของลำดับ CAG ในเซลล์ร่างกายมีส่วนเกี่ยวข้องกับความก้าวหน้าของโรค ในช่วงหลายทศวรรษ ยีน HTT จะเห็นลำดับ CAG ขยายตัวเป็นประมาณ 80 ชุด โดยตำแหน่ง CAG 35+ ทำให้เกิดข้อผิดพลาดในการเลื่อนเพิ่มเติมที่ขยายลำดับซ้ำ จากนั้นกระบวนการจะเร่งตัวขึ้นจนถึง 150 ชุดภายในไม่กี่ปี จะไม่มีผลกระทบที่เป็นพิษอย่างมีนัยสำคัญต่อเซลล์จนกว่าจะถึง 150 ชุด ณ จุดนั้น ยีนจำนวนมากจะทำงานผิดปกติมากขึ้นเรื่อยๆ ในช่วงหลายเดือนเซลล์ประสาทหนามขนาดกลางจะค่อยๆ สูญเสียเอกลักษณ์ของเซลล์ไปจนกระทั่งกลไกการตายของเซลล์ถูกกระตุ้น[ 59 ]
การเปลี่ยนแปลงในระดับมหภาค

ในขั้นต้น ความเสียหายต่อสมองจะมีความเฉพาะเจาะจงตามภูมิภาค โดยส่วนดอร์ซัลสไตรเอตัมในฐานสมองส่วนซับคอร์เทกซ์จะได้รับผลกระทบเป็นหลัก ตามมาด้วย การมีส่วนเกี่ยวข้อง ของคอร์เทกซ์ในทุกพื้นที่ ในภายหลัง [ 60 ] [ 61 ]พื้นที่อื่นๆ ของ ฐานสมอง ที่ได้รับผลกระทบ ได้แก่ ซับสแตนเซียไน ก รา การมีส่วนเกี่ยวข้องของคอร์เทกซ์ ได้แก่ชั้นคอร์เทกซ์ 3, 5 และ 6นอกจากนี้ยังเห็นได้ชัดว่ามีการมีส่วนเกี่ยวข้องของฮิปโปแคมปัสเซลล์พูร์คินเจในซีรีเบลลัมนิวเคลียสทูเบอรัลด้านข้างของไฮโปทาลามัสและบางส่วนของทาลามัส [ 29 ] พื้นที่เหล่านี้ได้รับผลกระทบตามโครงสร้างและชนิดของเซลล์ประสาทที่ประกอบอยู่ โดยขนาดจะลดลงเมื่อสูญเสียเซลล์[ 29 ]เซลล์ประสาทสไปนีขนาดกลางในส ไตรเอตัม มีความเปราะบางมากที่สุด โดยเฉพาะอย่างยิ่งเซลล์ที่มีการฉายภาพไปยังโกลบัสพัลลิดัสภายนอกในขณะที่ เซลล์ ประสาทอินเตอร์นิวรอนและเซลล์สไปนีที่ฉายภาพไปยังโกลบัสพัลลิดัสภายในได้รับผลกระทบน้อยกว่า[ 29 ] [ 62 ] HD ยังทำให้เกิดการเพิ่มขึ้นอย่างผิดปกติของแอสโทรไซต์และการกระตุ้นเซลล์ภูมิคุ้มกันของสมองไมโครเกลีย[ 63 ]
ฐานสมองมีบทบาทสำคัญในการควบคุมการเคลื่อนไหวและพฤติกรรม หน้าที่ของฐานสมองยังไม่เป็นที่เข้าใจอย่างถ่องแท้ แต่ทฤษฎีต่างๆ เสนอว่าฐานสมองเป็นส่วนหนึ่งของระบบบริหารจัดการ ทางปัญญา [ 31 ]และวงจรการเคลื่อนไหว[ 64 ]โดยปกติฐานสมองจะยับยั้งวงจรจำนวนมากที่สร้างการเคลื่อนไหวเฉพาะ เพื่อเริ่มต้นการเคลื่อนไหวเฉพาะอย่าง เปลือกสมองจะส่งสัญญาณไปยังฐานสมองซึ่งทำให้การยับยั้งถูกปลดปล่อย ความเสียหายต่อฐานสมองอาจทำให้การปลดปล่อยหรือการคืนสภาพของการยับยั้งเป็นไปอย่างไม่แน่นอนและควบคุมไม่ได้ ซึ่งส่งผลให้การเริ่มต้นการเคลื่อนไหวดูเก้งก้าง หรือการเคลื่อนไหวเริ่มต้นโดยไม่ตั้งใจ หรือการเคลื่อนไหวหยุดลงก่อนหรือเกินกว่าที่ตั้งใจไว้ ความเสียหายที่สะสมในบริเวณนี้ทำให้เกิดการเคลื่อนไหวที่ผิดปกติซึ่งเป็นลักษณะเฉพาะของโรคฮันติงตัน เรียกว่า โรคชักกระตุก (chorea ) ซึ่งเป็นภาวะการเคลื่อนไหวผิดปกติชนิดหนึ่ง [ 64 ]เนื่องจากความไม่สามารถของฐานสมองในการยับยั้งการเคลื่อนไหว ผู้ที่ได้รับผลกระทบจึงประสบกับความสามารถในการพูดและการกลืนอาหารและของเหลวที่ลดลงอย่างหลีกเลี่ยงไม่ได้ (ภาวะกลืนลำบาก) [ 65 ]
การควบคุมการถอดรหัสที่ผิดปกติ
โปรตีนที่จับกับ CREB (CBP) ซึ่งเป็นตัวควบคุมการถอดรหัสร่วม มีความสำคัญต่อการทำงานของเซลล์ เนื่องจากทำหน้าที่เป็นตัวกระตุ้นร่วมที่โปรโมเตอร์จำนวนมาก โดยจะกระตุ้นการถอดรหัสของยีนสำหรับวิถีการอยู่รอด[ 57 ] CBP มี โดเมน อะเซทิลทรานสเฟอ เรส ซึ่ง HTT จับผ่านโดเมนที่มีโพลีกลูตามีน[ 66 ]สมองที่ชันสูตรศพของผู้ที่เป็นโรคฮันติงตันยังพบว่ามีปริมาณ CBP ลดลงอย่างมาก[ 67 ]นอกจากนี้ เมื่อมีการแสดงออกของ CBP มากเกินไป การตายที่เกิดจากโพลีกลูตามีนจะลดลง ซึ่งแสดงให้เห็นเพิ่มเติมว่า CBP มีบทบาทสำคัญในโรคฮันติงตันและเซลล์ประสาทโดยทั่วไป[ 57 ]
การวินิจฉัย
การวินิจฉัยโรค HD สามารถทำได้เมื่อมีอาการทางกายภาพที่เฉพาะเจาะจงกับโรค[ 29 ]การตรวจทางพันธุกรรมสามารถใช้เพื่อยืนยันการวินิจฉัยทางกายภาพได้หากไม่มีประวัติครอบครัวเป็นโรค HD แม้ก่อนที่อาการจะเริ่มปรากฏ การตรวจทางพันธุกรรมก็สามารถยืนยันได้ว่าบุคคลหรือตัวอ่อนมีสำเนาของลำดับนิวคลีโอไทด์สามตัว (CAG) ที่ขยายตัวใน ยีน HTTที่ก่อให้เกิดโรคหรือไม่ มีบริการให้ คำปรึกษาทางพันธุกรรมเพื่อให้คำแนะนำและแนวทางตลอดขั้นตอนการตรวจและเกี่ยวกับผลกระทบของการวินิจฉัยที่ได้รับการยืนยัน ผลกระทบเหล่านี้รวมถึงผลกระทบต่อสภาพจิตใจ อาชีพ การตัดสินใจวางแผนครอบครัว ญาติ และความสัมพันธ์ของบุคคลนั้นๆ จากการศึกษาในปี 2550 พบว่าแม้จะมีการตรวจก่อนมีอาการ แต่มีเพียง 5% ของผู้ที่มีความเสี่ยงที่จะได้รับโรค HD ทางพันธุกรรมเท่านั้นที่เลือกที่จะทำการตรวจ[ 29 ]
ทางคลินิก

การตรวจร่างกายบางครั้งอาจรวมกับการตรวจทางจิตวิทยาสามารถช่วยระบุได้ว่าโรคได้เริ่มขึ้นแล้วหรือไม่[ 29 ] การเคลื่อนไหวโดยไม่ตั้งใจมากเกินไปของส่วนใดส่วนหนึ่งของร่างกาย มักเป็นสาเหตุที่ทำให้ต้องไปพบแพทย์ หากการเคลื่อนไหวเหล่านี้เกิดขึ้นอย่างกะทันหัน มีจังหวะเวลาและการกระจายตัวแบบสุ่ม ก็อาจบ่งชี้ถึงการวินิจฉัยโรคฮันติงตันได้ อาการทางด้านการรับรู้หรือพฤติกรรมนั้น มักไม่ใช่อาการแรกที่ได้รับการวินิจฉัย มักจะตรวจพบได้ในภายหลังหรือเมื่ออาการพัฒนาขึ้นไปอีกขั้น ความรุนแรงของโรคสามารถวัดได้โดยใช้มาตราส่วนการให้คะแนนโรคฮันติงตันแบบรวม ซึ่งเป็นระบบการให้คะแนนโดยรวมโดยอิงจากการประเมินด้านการเคลื่อนไหว พฤติกรรม การรับรู้ และการทำงาน[ 69 ] [ 70 ]การถ่ายภาพทางการแพทย์เช่นการสแกน CTหรือการสแกน MRIสามารถแสดงให้เห็นการฝ่อของนิวเคลียสคอเดตในช่วงเริ่มต้นของโรค ดังที่เห็นในภาพประกอบทางด้านขวา แต่การเปลี่ยนแปลงเหล่านี้เพียงอย่างเดียวไม่ถือเป็นการวินิจฉัยโรคฮันติงตันการฝ่อของสมองสามารถพบได้ในระยะขั้นสูงของโรค เทคนิค การสร้างภาพประสาทเชิงฟังก์ชันเช่นการสร้างภาพด้วยคลื่นแม่เหล็กไฟฟ้าเชิงฟังก์ชัน (fMRI) และการสร้างภาพด้วยโพซิตรอน (PET) สามารถแสดงการเปลี่ยนแปลงในกิจกรรมของสมองก่อนที่จะเกิดอาการทางกายภาพ แต่เป็นเครื่องมือทดลองและไม่ได้นำมาใช้ในทางคลินิก[ 29 ]
การตรวจทางพันธุกรรมเพื่อการทำนายผล
เนื่องจาก HD มีรูปแบบการถ่ายทอดทางพันธุกรรมแบบออโตโซมัลโดมิแนนต์ จึงมีแรงจูงใจอย่างมากสำหรับบุคคลที่มีความเสี่ยงที่จะได้รับโรคนี้ในการแสวงหาการวินิจฉัย การทดสอบทางพันธุกรรมสำหรับ HD ประกอบด้วยการตรวจเลือด ซึ่งจะนับจำนวนการทำซ้ำของ CAG ในแต่ละอัลลีลHTT [ 71 ]ค่าเกณฑ์กำหนดไว้ดังนี้:
- เมื่อมีการทำซ้ำ CAG 40 ครั้งขึ้นไป จะพบอัลลีล ที่มีการแสดงออก เต็มที่ (FPA) [ 72 ] โดยทั่วไปแล้ว " การทดสอบที่เป็นบวก " หรือ "ผลลัพธ์ที่เป็นบวก" หมายถึงกรณีนี้ ผลลัพธ์ที่เป็นบวกไม่ได้ถือเป็นการวินิจฉัย เนื่องจากอาจได้รับผลการทดสอบหลายสิบปีก่อนที่อาการจะเริ่มปรากฏ อย่างไรก็ตาม การทดสอบที่เป็นลบหมายความว่าบุคคลนั้นไม่มียีนที่ขยายตัวและจะไม่เป็นโรค HD [ 29 ]การทดสอบจะบอกบุคคลที่เดิมมีโอกาส 50% ที่จะได้รับโรคนี้ว่าความเสี่ยงของพวกเขาเพิ่มขึ้นเป็น 100% หรือหมดไป บุคคลที่ตรวจพบว่าเป็นโรคนี้จะเป็นโรค HD ในช่วงใดช่วงหนึ่งของชีวิต หากพวกเขามีชีวิตอยู่ได้นานพอที่อาการจะปรากฏ[ 29 ]
- ที่จำนวนการทำซ้ำ 36 ถึง 39 ครั้ง อัลลีลที่มีการแสดงออกไม่สมบูรณ์หรือลดลง (RPA) อาจทำให้เกิดอาการ โดยปกติจะเกิดขึ้นในวัยผู้ใหญ่ตอนปลาย[ 72 ]ความเสี่ยงสูงสุดคือ 60% ที่บุคคลที่มี RPA จะมีอาการเมื่ออายุ 65 ปี และ 70% เมื่ออายุ 75 ปี[ 72 ]
- ที่จำนวนการทำซ้ำ 27 ถึง 35 ครั้ง อัลลีลระดับกลาง (IA) หรืออัลลีลปกติขนาดใหญ่ ไม่เกี่ยวข้องกับโรคที่มีอาการในบุคคลที่ได้รับการทดสอบ แต่อาจขยายตัวเมื่อมีการถ่ายทอดทางพันธุกรรมต่อไปจนทำให้เกิดอาการในลูกหลานได้[ 72 ]
- หากมีการทำซ้ำ 26 ครั้งหรือน้อยกว่า ผลลัพธ์จะไม่เกี่ยวข้องกับ HD [ 72 ]
การทดสอบก่อนเริ่มมีอาการถือเป็นเหตุการณ์ที่เปลี่ยนแปลงชีวิตและเป็นการตัดสินใจส่วนตัวอย่างมาก[ 29 ]เหตุผลหลักที่ให้ไว้สำหรับการเลือกทดสอบ HD คือเพื่อช่วยในการตัดสินใจด้านอาชีพและครอบครัว[ 29 ]การทดสอบทำนายโรคฮันติงตันมีให้บริการผ่านการวิเคราะห์การเชื่อมโยง (ซึ่งต้องทดสอบสมาชิกในครอบครัวหลายคน) ตั้งแต่ปี 1986 และผ่านการวิเคราะห์การกลายพันธุ์โดยตรงตั้งแต่ปี 1993 [ 73 ]ในเวลานั้น การสำรวจระบุว่า 50–70% ของบุคคลที่มีความเสี่ยงจะสนใจรับการทดสอบ แต่หลังจากมีการเสนอการทดสอบทำนายแล้ว มีคนจำนวนน้อยลงที่เลือกรับการทดสอบ[ 74 ]มากกว่า 95% ของบุคคลที่มีความเสี่ยงที่จะได้รับ HD ทางพันธุกรรมไม่ได้ดำเนินการทดสอบ ส่วนใหญ่เป็นเพราะไม่มีวิธีการรักษา[ 29 ]ประเด็นสำคัญคือความวิตกกังวลที่บุคคลประสบเกี่ยวกับการไม่รู้ว่าพวกเขาจะพัฒนาเป็น HD ในที่สุดหรือไม่ เมื่อเทียบกับผลกระทบของผลลัพธ์ที่เป็นบวก[ 29 ]ไม่ว่าผลลัพธ์จะเป็นอย่างไร ระดับความเครียดจะลดลงสองปีหลังจากได้รับการทดสอบ แต่ความเสี่ยงต่อการฆ่าตัวตายจะเพิ่มขึ้นหลังจากผลการทดสอบเป็นบวก[ 29 ]บุคคลที่พบว่าไม่ได้รับมรดกความผิดปกติอาจรู้สึกผิดที่รอดชีวิตจากสมาชิกในครอบครัวที่ได้รับผลกระทบ[ 29 ]ปัจจัยอื่นๆ ที่นำมาพิจารณาเมื่อทำการทดสอบ ได้แก่ ความเป็นไปได้ของการเลือกปฏิบัติและผลกระทบของผลลัพธ์ที่เป็นบวก ซึ่งโดยปกติหมายความว่าพ่อแม่มียีนที่ได้รับผลกระทบ และพี่น้องของบุคคลนั้นจะมีความเสี่ยงที่จะได้รับมรดกยีนนั้น[ 29 ]ในการศึกษาหนึ่ง พบว่ามีการเลือกปฏิบัติทางพันธุกรรมใน 46% ของบุคคลที่มีความเสี่ยงต่อโรคฮันติงตัน เกิดขึ้นในอัตราที่สูงกว่าในความสัมพันธ์ส่วนตัวมากกว่าในประกันสุขภาพหรือความสัมพันธ์ในการจ้างงาน[ 75 ]การให้คำปรึกษาทางพันธุกรรมใน HD สามารถให้ข้อมูล คำแนะนำ และการสนับสนุนสำหรับการตัดสินใจเบื้องต้น และหากเลือกแล้ว ตลอดทุกขั้นตอนของกระบวนการทดสอบ[ 76 ]เนื่องจากผลกระทบของการทดสอบนี้ ผู้ป่วยที่ต้องการเข้ารับการทดสอบจะต้องเข้ารับการให้คำปรึกษา 3 ครั้ง ซึ่งจะให้ข้อมูลเกี่ยวกับโรคฮันติงตัน[ 77 ]
การให้คำปรึกษาและแนวทางการใช้การตรวจทางพันธุกรรมสำหรับ HD ได้กลายเป็นแบบอย่างสำหรับความผิดปกติทางพันธุกรรมอื่นๆ เช่นโรคสมองน้อยเสื่อมแบบถ่ายทอด ทางพันธุกรรมแบบเด่น [ 29 ] [ 78 ] [ 79 ]การตรวจก่อนมีอาการของ HD ยังมีอิทธิพลต่อการตรวจโรคอื่นๆ ที่มีการกลายพันธุ์ทางพันธุกรรม เช่น โรค ไตถุงน้ำหลายถุงโรคอัลไซเมอร์ในครอบครัวและมะเร็งเต้านม[ 78 ] เครือข่ายคุณภาพพันธุศาสตร์โมเลกุลแห่งยุโรป ได้เผยแพร่แผนการประเมินคุณภาพภายนอกประจำปีสำหรับการตรวจทางพันธุกรรมโมเลกุลสำหรับโรคนี้ และได้พัฒนาแนวทางปฏิบัติที่ดีที่สุดสำหรับการตรวจทางพันธุกรรมสำหรับ HD เพื่อช่วยในการตรวจและรายงานผล[ 80 ]
การวินิจฉัยทางพันธุกรรมก่อนการฝังตัวของตัวอ่อน
ตัวอ่อนที่ได้จากการผสมเทียมในหลอดทดลองสามารถตรวจทางพันธุกรรมเพื่อหาความเสี่ยงต่อโรคฮันติงตัน (HD) ได้โดยใช้การวินิจฉัยทางพันธุกรรมก่อนการฝังตัว (Preimplantation Genetic Diagnosis: PGE ) เทคนิคนี้ คือการสกัดเซลล์หนึ่งหรือสองเซลล์จากตัวอ่อนที่มีเซลล์ประมาณ 4-8 เซลล์ แล้วนำไปทดสอบหาความผิดปกติทางพันธุกรรม จากนั้นจึงใช้เทคนิคนี้เพื่อให้แน่ใจว่าตัวอ่อนที่มียีน HD จะไม่ถูกฝังตัวในมดลูก เพื่อไม่ให้ลูกหลานได้รับโรคนี้ การวินิจฉัยทางพันธุกรรมก่อนการฝังตัวบางรูปแบบ—เช่น การตรวจแบบไม่เปิดเผยข้อมูลหรือการตรวจเพื่อตัดความเสี่ยง—ช่วยให้ผู้ที่มีความเสี่ยงสามารถมีลูกหลานที่ปราศจากโรค HD ได้โดยไม่ต้องเปิดเผยข้อมูลทางพันธุกรรมของพ่อแม่ ทำให้ไม่มีข้อมูลว่าตนเองมีความเสี่ยงที่จะเป็นโรค HD หรือไม่ ในการตรวจเพื่อตัดความเสี่ยงนั้น จะมีการเปรียบเทียบดีเอ็นเอของตัวอ่อนกับดีเอ็นเอของพ่อแม่และปู่ย่าตายาย เพื่อหลีกเลี่ยงการถ่ายทอดบริเวณโครโมโซมที่มียีน HD จากปู่ย่าตายายที่เป็นโรค ในการตรวจแบบไม่เปิดเผยข้อมูลนั้น จะมีการนำเฉพาะตัวอ่อนที่ปราศจากโรคกลับไปฝังตัวในมดลูกเท่านั้น โดยจะไม่เปิดเผยข้อมูลทางพันธุกรรมของพ่อแม่และความเสี่ยงต่อโรค HD ของพ่อแม่เลย[ 81 ] [ 82 ]
การตรวจก่อนคลอด
การวินิจฉัยก่อนคลอดสำหรับตัวอ่อนหรือทารกในครรภ์ก็เป็นไปได้เช่นกัน โดยใช้สารพันธุกรรมของทารกในครรภ์ที่ได้จากการเก็บตัวอย่างรกการเจาะน้ำคร่ำสามารถทำได้หากการตั้งครรภ์ดำเนินไปได้ระยะหนึ่ง คือภายใน 14–18 สัปดาห์ ขั้นตอนนี้จะตรวจสอบน้ำคร่ำที่อยู่รอบทารกเพื่อหาตัวบ่งชี้ของการกลายพันธุ์ของ HD [ 83 ]วิธีนี้ยังสามารถใช้ร่วมกับการทดสอบการยกเว้นเพื่อหลีกเลี่ยงการเปิดเผยจีโนไทป์ของพ่อแม่ การทดสอบก่อนคลอดสามารถทำได้เมื่อพ่อแม่ได้รับการวินิจฉัยว่าเป็น HD เมื่อพวกเขาได้รับการทดสอบทางพันธุกรรมที่แสดงให้เห็นถึงการขยายตัวของ ยีน HTTหรือเมื่อพวกเขามีโอกาส 50% ที่จะได้รับโรคนี้ พ่อแม่สามารถได้รับคำแนะนำเกี่ยวกับทางเลือกต่างๆ ซึ่งรวมถึงการยุติการตั้งครรภ์และเกี่ยวกับความยากลำบากของเด็กที่มียีนที่ระบุ[ 84 ] [ 85 ]
นอกจากนี้ ในการตั้งครรภ์ที่มีความเสี่ยงเนื่องจากคู่ครองชายได้รับผลกระทบ การวินิจฉัยก่อนคลอดแบบไม่รุกรานสามารถทำได้โดยการวิเคราะห์ดีเอ็นเอของทารกในครรภ์ที่ลอยอยู่ในเลือดจากตัวอย่างเลือดที่เก็บจากมารดา (ผ่านการเจาะเลือด ) ระหว่างสัปดาห์ที่ 6 ถึง 12 ของการตั้งครรภ์[ 72 ]ซึ่งไม่มีความเสี่ยงต่อการแท้งบุตรที่เกี่ยวข้องกับขั้นตอน[ 72 ]
การวินิจฉัยแยกโรค
ประมาณ 99% ของการวินิจฉัย HD โดยอาศัยอาการทั่วไปและประวัติครอบครัวของโรคได้รับการยืนยันโดยการทดสอบทางพันธุกรรมว่ามีการขยายตัวของลำดับนิวคลีโอไทด์สามตัวที่ทำให้เกิด HD ส่วนที่เหลือส่วนใหญ่เรียกว่ากลุ่มอาการคล้าย HD (HDL) [ 29 ] [ 86 ]สาเหตุของโรค HDL ส่วนใหญ่ยังไม่ทราบ แต่โรคที่มีสาเหตุที่ทราบนั้นเกิดจากการกลายพันธุ์ในยีนโปรตีนพรีออน (HDL1) ยีนจังก์โทฟิลิน 3 (HDL2) ยีนที่ไม่ทราบที่ถ่ายทอดแบบด้อย (HDL3—พบในสองครอบครัวเท่านั้นและยังไม่เป็นที่เข้าใจอย่างถ่องแท้) และยีนที่เข้ารหัสโปรตีนที่จับกับกล่อง TATA ( SCA17 บางครั้งเรียกว่า HDL4 ) โรคที่ถ่ายทอดแบบออโตโซมัลเด่นอื่นๆ ที่อาจวินิจฉัยผิดว่าเป็น HD ได้แก่ โรคสมองฝ่อเดน ทาโทรูบรัล-พัลลิโดลูยเซียนและ โรค นิวโรเฟอร์ริติโนพา ธี นอกจาก นี้ ความผิดปกติที่ถ่ายทอดแบบออโตโซมัลด้อยบางอย่างก็คล้ายกับกรณีที่เกิดขึ้นเองของ HD ซึ่งรวมถึง โรค อะแคนโทไซโตซิสแบบโคเรียและโรคความเสื่อมของระบบประสาทที่เกี่ยวข้องกับแพนโทเทเนตไคเนส โรค ที่ถ่ายทอดทางโครโมโซม X ชนิด หนึ่งคือโรคแมคเลียดซินโดรม[ 86 ]
การจัดการ

มีวิธีการรักษาเพื่อลดความรุนแรงของอาการ HD บางอย่าง[ 87 ]สำหรับการรักษาเหล่านี้จำนวนมาก หลักฐานที่ยืนยันประสิทธิภาพในการรักษาอาการของ HD โดยเฉพาะยังไม่สมบูรณ์[ 29 ] [ 88 ]เมื่อโรคดำเนินไป ความสามารถในการดูแลตนเองจะลดลง และการดูแลแบบสหสาขาวิชาชีพที่ ได้รับการจัดการอย่างระมัดระวัง จึงมีความจำเป็นมากขึ้น[ 29 ] แม้ว่าจะมีงานวิจัยเกี่ยวกับการออกกำลังกายและการบำบัดที่แสดงให้เห็นว่ามีประโยชน์ใน การฟื้นฟู อาการทางด้านความรู้ความเข้าใจของ HD ค่อนข้างน้อย แต่หลักฐานบางอย่างแสดงให้เห็นถึงประโยชน์ของการบำบัดทางกายภาพการบำบัดทางอาชีพและการบำบัดด้านการพูด[ 29 ]
การบำบัด
การลดน้ำหนักและปัญหาในการรับประทานอาหารเนื่องจากภาวะกลืนลำบากและความผิดปกติของกล้ามเนื้ออื่นๆ เป็นเรื่องปกติ ทำให้การจัดการด้านโภชนาการมีความสำคัญมากขึ้นเมื่อโรคดำเนินไป[ 29 ] สามารถเติมสารเพิ่มความข้นลง ในของเหลวได้ เนื่องจากของเหลวที่ข้นกว่าจะกลืนได้ง่ายและปลอดภัยกว่า [ 29 ]การเตือนผู้ป่วยให้รับประทานอาหารช้าๆ และนำอาหารชิ้นเล็กๆ เข้าปากก็อาจช่วยป้องกันการสำลักได้เช่นกัน[ 29 ]หากการรับประทานอาหารกลายเป็นอันตรายหรือไม่สะดวกสบายเกินไปก็มี ทางเลือกในการใช้ท่อ ให้อาหารทางกระเพาะอาหารแบบส่องกล้องผ่านผิวหนัง ท่อให้อาหารนี้จะติดอยู่ถาวรผ่านทาง ช่องท้องเข้าไปในกระเพาะ อาหาร ช่วย ลดความเสี่ยงของการสำลักอาหารและให้การจัดการด้านโภชนาการที่ดีขึ้น[ 89 ] แนะนำให้ ทำการประเมินและจัดการโดยนักพยาธิวิทยาด้านการพูดและภาษาที่มีประสบการณ์ในโรคฮันติงตัน[ 29 ]
ผู้ป่วยโรคฮันติงตันอาจพบนักกายภาพบำบัดเพื่อจัดการกับอาการทางกายภาพด้วยวิธีการที่ไม่ต้องผ่าตัดและไม่ใช้ยา นักกายภาพบำบัดอาจทำการประเมินและป้องกันความเสี่ยงต่อการหกล้ม รวมถึงการเสริมสร้างความแข็งแรง การยืดกล้ามเนื้อ และการออกกำลังกายแบบคาร์ดิโอ อาจมีการสั่งจ่าย อุปกรณ์ช่วยเดินตามความเหมาะสม นอกจากนี้ นักกายภาพบำบัดยังสั่งจ่ายการฝึกหายใจและเทคนิคการเคลียร์ทางเดินหายใจเมื่อมีปัญหาเกี่ยวกับระบบทางเดินหายใจ[ 90 ]เครือข่าย HD ของยุโรปได้จัดทำแนวทางปฏิบัติที่เป็นเอกฉันท์เกี่ยวกับการกายภาพบำบัดในโรคฮันติงตัน[ 90 ]เป้าหมายของการฟื้นฟูสมรรถภาพในระยะเริ่มต้นคือการป้องกันการสูญเสียการทำงาน การเข้าร่วมโปรแกรมฟื้นฟูสมรรถภาพในช่วงระยะเริ่มต้นถึงระยะกลางของโรคอาจเป็นประโยชน์ เนื่องจากจะนำไปสู่การรักษาสมรรถภาพการเคลื่อนไหวและการทำงานในระยะยาว การฟื้นฟูสมรรถภาพในช่วงระยะท้ายมีจุดมุ่งหมายเพื่อชดเชยการสูญเสียการเคลื่อนไหวและการทำงาน[ 91 ]สำหรับการจัดการตนเองในระยะยาว นักกายภาพบำบัดอาจพัฒนาโปรแกรมการออกกำลังกายที่บ้านสำหรับผู้ป่วยที่เหมาะสม[ 92 ]
การดูแลแบบประคับประคองอาจช่วยปรับปรุงคุณภาพชีวิตของผู้ป่วยที่เป็นโรค HD ได้ด้วยการรักษาอาการและความเครียดทางจิตใจจากการเป็นโรคเสื่อมที่ดำเนินไปอย่างต่อเนื่อง[ 93 ]
ยา

Tetrabenazineได้รับการอนุมัติในปี 2000 สำหรับการรักษาอาการชักกระตุกในโรคฮันติงตันในสหภาพยุโรป และในปี 2008 ในสหรัฐอเมริกา[ 94 ]แม้ว่าจะมียาอื่นๆ ที่ใช้ " นอกเหนือข้อบ่งใช้ " แต่ tetrabenazine ก็เป็นยาตัวแรกที่ได้รับการอนุมัติสำหรับการรักษาโรคฮันติงตันในสหรัฐอเมริกา สารประกอบนี้เป็นที่รู้จักมาตั้งแต่ทศวรรษ 1950 ในปี 2017 deutetrabenazineซึ่งเป็นยา tetrabenazine รูปแบบที่หนักกว่าสำหรับการรักษาอาการชักกระตุกใน HD ได้รับการอนุมัติจาก FDA [ 95 ]ยานี้วางจำหน่ายในชื่อ Austedo Valbenazine (Ingrezza)ก็ได้รับการอนุมัติจาก FDA สำหรับการรักษาอาการชักกระตุกในโรคฮันติงตันในปี 2023 เช่นกัน[ 96 ] Tetrabenazine , deutetrabenazineและvalbenazineล้วนเป็น สารยับยั้ง vesicular monoamine transporter 2 (VMAT2)ซึ่งทำงานโดยการลดโดปามีนในสมอง ลดการเคลื่อนไหวโดยไม่ตั้งใจ[ 97 ]ยาเหล่านี้เป็นยาเพียงชนิดเดียวที่ได้รับการอนุมัติสำหรับโรคฮันติงตันโดยเฉพาะ (โดยเฉพาะอาการชักกระตุกที่เกี่ยวข้องกับโรคนี้)
ยาอื่นๆ ที่ช่วยลดอาการชักกระตุก ได้แก่ยาต้านโรคจิตและเบนโซไดอะซีพีน [ 26 ] อาการเคลื่อนไหวช้าและอาการแข็งเกร็ง โดยเฉพาะในกรณีเด็กและเยาวชน สามารถรักษาได้ด้วย ยา ต้านโรคพาร์ กินสัน และ อาการเคลื่อนไหว เร็วแบบไมโอโคลนิกสามารถรักษาได้ด้วยกรดวาลโปรอิก [ 26 ] หลักฐานเบื้องต้นพบว่ากรดเอทิลไอโคซาเพนตาอีโนอิกช่วยปรับปรุงอาการทางมอเตอร์ได้ภายในหนึ่งปี[ 98 ]อะแมนทาดีนก็ถูกนำมาใช้รักษาอาการชักกระตุกเช่นกัน แต่มีหลักฐานจำกัดเกี่ยวกับความปลอดภัยและประสิทธิภาพของยา[ 99 ]
อาการทางจิตเวชสามารถรักษาได้ด้วยยาที่คล้ายกับที่ใช้ในประชากรทั่วไป[ 29 ] [ 88 ]สารยับยั้งการดูดซึมเซโรโทนินแบบเลือกและ เมอร์ ทาซาพีนได้รับการแนะนำสำหรับภาวะซึมเศร้า ในขณะที่ยาต้านโรคจิตผิดปกติได้รับการแนะนำสำหรับโรคจิตและปัญหาพฤติกรรม[ 88 ]แนะนำให้ปรึกษาผู้เชี่ยวชาญด้านประสาทจิตเวช เนื่องจากผู้ป่วยอาจต้องได้รับการรักษาในระยะยาวด้วยยาหลายชนิดร่วมกัน[ 29 ]
ยาที่ทำจากพืช
There has been a number of alternative therapies experimented in ayurvedic medicine with plant-based products, although none have provided good evidence of efficacy. A recent study showed that the stromal processing peptidase (SPP), a synthetic enzyme found in plant chloroplasts, prevented the aggregation of proteins associated with Huntington's disease.[100] However, repeat studies and clinical validation are needed to confirm its true therapeutic potential.
Education
The families of individuals, and society at large, who have inherited or are at risk of inheriting HD have generations of experience of HD but may be unaware of recent breakthroughs in understanding the disease, and of the availability of genetic testing. Genetic counseling benefits these individuals by updating their knowledge, seeking to dispel any unfounded beliefs that they may have, and helping them consider their future options and plans. The Patient Education Program for Huntington's Disease has been created to help educate family members, caretakers, and those diagnosed with Huntington's disease.[101] Also covered is information concerning family planning choices, care management, and other considerations.[29][102]
Prognosis
The length of the trinucleotide repeat accounts for 60% of the variation of the age of symptoms onset and their rate of progress. A longer repeat results in an earlier age of onset and a faster progression of symptoms.[29][103] Individuals with more than sixty repeats often develop the disease before age 20, while those with fewer than 40 repeats may remain asymptomatic.[104] The remaining variation is due to environmental factors and other genes that influence the mechanism of the disease.[29]
โดยทั่วไปแล้ว อายุขัยของผู้ป่วยโรคฮันติงตันจะอยู่ที่ประมาณ 15 ถึง 20 ปีหลังจากเริ่มมีอาการที่มองเห็นได้[ 29 ]โรคฮันติงตันในวัยเด็กมีอายุขัยเฉลี่ย 10 ปีหลังจากเริ่มมีอาการที่มองเห็นได้ ผู้ป่วยโรคฮันติงตันมักจะมีอายุขัย เฉลี่ยลดลง เหลือประมาณ 58–63 ปี (ค่าเฉลี่ย) แม้ว่าจะแตกต่างกันอย่างมาก และการศึกษาต่างๆ ก็ให้ช่วงอายุที่แตกต่างกันเล็กน้อย[ 105 ] [ 106 ] [ 107 ]ภาวะแทรกซ้อนที่คุกคามชีวิตส่วนใหญ่เกิดจากการประสานงานของกล้ามเนื้อ และในระดับที่น้อยกว่าคือการเปลี่ยนแปลงพฤติกรรมที่เกิดจากการทำงานของสมองที่ลดลง ความเสี่ยงที่ใหญ่ที่สุดคือโรคปอดบวมซึ่งเป็นสาเหตุของการเสียชีวิตในผู้ป่วยโรคฮันติงตันหนึ่งในสาม เมื่อความสามารถในการประสานการเคลื่อนไหวเสื่อมลง ความยากลำบากในการกำจัดเสมหะออกจากปอด และความเสี่ยงที่เพิ่มขึ้นของการสำลักอาหารหรือเครื่องดื่ม ล้วนเพิ่มความเสี่ยงต่อการเป็นโรคปอดบวมความเสี่ยงที่มากเป็นอันดับสองคือโรคหัวใจซึ่งเป็นสาเหตุของการเสียชีวิตเกือบหนึ่งในสี่ของผู้ป่วยโรคฮันติงตัน[ 108 ]การฆ่าตัวตายเป็นสาเหตุการเสียชีวิตอันดับสาม โดย 7.3% ของผู้ป่วยที่เป็นโรค HD ฆ่าตัวตาย และมากถึง 27% พยายามฆ่าตัวตาย ยังไม่ชัดเจนว่าความคิดฆ่าตัวตายได้รับอิทธิพลจากอาการทางพฤติกรรมมากน้อยเพียงใด เนื่องจากอาการเหล่านี้บ่งบอกถึงความปรารถนาที่จะหลีกเลี่ยงระยะหลังของโรค[ 109 ] [ 110 ] [ 111 ]การฆ่าตัวตายเป็นความเสี่ยงสูงสุดของโรคนี้ก่อนที่จะได้รับการวินิจฉัย และในระยะกลางของการพัฒนาของโรค ความเสี่ยงอื่นๆ ที่เกี่ยวข้อง ได้แก่ การสำลักเนื่องจากไม่สามารถกลืนได้การบาดเจ็บทางร่างกายจากการหกล้ม และภาวะทุพโภชนาการ[ 108 ] [ 22 ]
ระบาดวิทยา
โรคฮันติงตันมักเริ่มแสดงอาการช้า ทำให้โดยทั่วไปแล้วจะไม่ส่งผลกระทบต่อการสืบพันธุ์[ 29 ]อัตราการแพร่ระบาดทั่วโลกของโรคฮันติงตันอยู่ที่ 5-10 รายต่อประชากร 100,000 คน[ 112 ] [ 113 ]แต่แตกต่างกันอย่างมากในแต่ละพื้นที่ทางภูมิศาสตร์ อันเป็นผลมาจากเชื้อชาติ การย้ายถิ่นฐานในท้องถิ่น และรูปแบบการอพยพในอดีต[ 29 ]อัตราการแพร่ระบาดใกล้เคียงกันในผู้ชายและผู้หญิง อัตราการเกิดโรคสูงที่สุดในกลุ่มคนที่มีเชื้อสายยุโรปตะวันตก โดยเฉลี่ยประมาณ 7 รายต่อประชากร 100,000 คน และต่ำกว่าในส่วนอื่นๆ ของโลก เช่น 1 รายต่อประชากร 1 ล้านคนในกลุ่มคนที่มีเชื้อสายเอเชียและแอฟริกา การศึกษาทางระบาดวิทยาในปี 2013 เกี่ยวกับการแพร่ระบาดของโรคฮันติงตันในสหราชอาณาจักรระหว่างปี 1990 ถึง 2010 พบว่าอัตราการแพร่ระบาดเฉลี่ยของสหราชอาณาจักรอยู่ที่ 12.3 รายต่อประชากร 100,000 คน[ 29 ] [ 114 ]นอกจากนี้ บางพื้นที่เฉพาะที่ยังมีอัตราการแพร่ระบาดสูงกว่าค่าเฉลี่ยของภูมิภาคมาก[ 29 ]หนึ่งในพื้นที่ที่มีอัตราการเกิดโรคสูงที่สุดคือในประชากรที่แยกตัวออกจากกันใน ภูมิภาค ทะเลสาบมาราไคโบของเวเนซุเอลาซึ่งโรค HD ส่งผลกระทบต่อประชากรมากถึง 700 คนต่อ 100,000 คน[ 29 ] [ 115 ]พื้นที่อื่นๆ ที่มีอัตราการแพร่ระบาดสูงพบได้ในแทสเมเนียและบางภูมิภาคของสกอตแลนด์เวลส์และสวีเดน[ 111 ]ในบางกรณี อัตราการแพร่ระบาดที่เพิ่มขึ้นเกิดจากปรากฏการณ์ผู้ก่อตั้ง ในท้องถิ่น ซึ่ง เป็นการอพยพของผู้ติดเชื้อในอดีตเข้ามาในพื้นที่ที่มีการแยกตัวทางภูมิศาสตร์[ 111 ] [ 116 ]ผู้ติดเชื้อบางรายสามารถสืบย้อนกลับไปได้หลายร้อยปีโดยใช้การศึกษาทางพันธุศาสตร์[ 111 ] แฮ ปโลไทป์ทางพันธุกรรมยังสามารถให้เบาะแสเกี่ยวกับความแปรผันทางภูมิศาสตร์ของอัตราการแพร่ระบาดได้อีกด้วย[ 111 ] [ 117 ]ในทางตรงกันข้ามไอซ์แลนด์ มีอัตราการแพร่ระบาดค่อนข้างต่ำเพียง 1 ต่อ 100,000 คน แม้ว่าชาว ไอซ์แลนด์จะสืบเชื้อสายมาจากชนเผ่าเยอรมันยุคแรกของสแกนดิเนเวียซึ่งเป็นต้นกำเนิดของชาวสวีเดน ก็ตาม โดยทุกกรณี ยกเว้นกรณีหนึ่งที่ย้อนกลับไปเกือบสองศตวรรษ ล้วนสืบเชื้อสายมาจากคู่สามีภรรยาที่อาศัยอยู่ในช่วงต้นศตวรรษที่ 19 [ 118 ]ฟินแลนด์นอกจากนี้ ยังมีอุบัติการณ์ต่ำเพียง 2.2 ต่อ 100,000 คน[ 119 ]
ก่อนการค้นพบการทดสอบทางพันธุกรรม สถิติสามารถรวมเฉพาะการวินิจฉัยทางคลินิกตามอาการทางกายภาพและประวัติครอบครัวของ HD เท่านั้น โดยไม่รวมผู้ที่เสียชีวิตจากสาเหตุอื่นก่อนได้รับการวินิจฉัย ปัจจุบันกรณีเหล่านี้สามารถรวมอยู่ในสถิติได้แล้ว และเมื่อการทดสอบแพร่หลายมากขึ้น การประมาณการความชุกและอุบัติการณ์ของโรคก็มีแนวโน้มที่จะเพิ่มขึ้น[ 111 ] [ 120 ]
ประวัติศาสตร์

ในอดีตหลายศตวรรษที่ผ่านมา โรคชักกระตุกชนิดต่างๆ บางครั้งถูกเรียกด้วยชื่อต่างๆ เช่นโรคเต้นรำของนักบุญวิตัสโดยที่แทบไม่มีความเข้าใจถึงสาเหตุหรือประเภทของโรคในแต่ละกรณีเลย
การกล่าวถึง HD อย่างชัดเจนครั้งแรกปรากฏในจดหมายของCharles Oscar Waters (1816–1892) ซึ่งตีพิมพ์ในฉบับพิมพ์ครั้งแรกของRobley Dunglison 's Practice of Medicineในปี 1842 [ 122 ] Waters อธิบายถึง "โรคชักกระตุกชนิดหนึ่ง ซึ่งเรียกกันทั่วไปว่า magrums" รวมถึงคำอธิบายที่ถูกต้องเกี่ยวกับโรคชักกระตุก ความก้าวหน้าของโรค และการถ่ายทอดทางพันธุกรรมของโรค[ 123 ]ในปี 1846 Charles Rollin Gorman (1817–1879) สังเกตว่าอัตราการเกิดโรคที่สูงขึ้นดูเหมือนจะเกิดขึ้นในภูมิภาคเฉพาะที่[ 124 ] [ 123 ]นอกเหนือจากกอร์แมนและวอเตอร์ส ซึ่งทั้งคู่เป็นนักศึกษาของดังกลิสันที่วิทยาลัยการแพทย์เจฟเฟอร์สันในฟิลาเดลเฟีย[ 125 ]โยฮัน คริสเตียน ลุนด์ (1830–1906) ยังได้เขียนคำอธิบายเบื้องต้นในปี 1860 [ 123 ]เขาระบุโดยเฉพาะว่าในเซเตสดาเลนหุบเขาบนภูเขาที่เงียบสงบในนอร์เวย์อัตราการเป็นภาวะสมองเสื่อมที่สูงนั้นเกี่ยวข้องกับรูปแบบของความผิดปกติของการเคลื่อนไหวแบบกระตุกที่ถ่ายทอดทางพันธุกรรม[ 126 ]
คำอธิบายอย่างละเอียดเกี่ยวกับโรคนี้ครั้งแรกนั้นมาจากจอร์จ ฮันติงตันในปี 1872 โดยการตรวจสอบประวัติทางการแพทย์ของคนหลายรุ่นในครอบครัวเดียวกันที่มีอาการคล้ายคลึงกัน เขาตระหนักว่าอาการเหล่านั้นต้องมีความเชื่อมโยงกัน เขาจึงนำเสนอคำจำกัดความที่ละเอียดและแม่นยำของโรคนี้ในบทความแรกของเขา ฮันติงตันได้อธิบายรูปแบบการถ่ายทอดทางพันธุกรรมแบบเด่นของโครโมโซมร่างกายอย่างแม่นยำหลายปีก่อนที่นักวิทยาศาสตร์จะค้นพบการถ่ายทอดทางพันธุกรรมแบบเมนเดล อีก ครั้ง
เป็นโรคที่ถ่ายทอดทางพันธุกรรม เมื่อพ่อหรือแม่ หรือทั้งสองคนมีอาการของโรค ... ลูกหลานอย่างน้อยหนึ่งคนมักจะป่วยเป็นโรคนี้ ... แต่ถ้าหากลูกหลานเหล่านี้รอดชีวิตโดยไม่เป็นโรคนี้ แสดงว่าสายสัมพันธ์ทางพันธุกรรมขาดลง และหลานและเหลนของผู้ที่เป็นโรคนี้แต่เดิมสามารถมั่นใจได้ว่าพวกเขาปลอดจากโรคนี้[ 121 ] [ 127 ]
เซอร์วิลเลียม ออสเลอร์สนใจในความผิดปกติและโรคชักกระตุกโดยทั่วไป และประทับใจกับบทความของฮันติงตัน โดยกล่าวว่า "ในประวัติศาสตร์การแพทย์ มีเพียงไม่กี่กรณีที่โรคได้รับการอธิบายอย่างแม่นยำ ชัดเจน หรือกระชับกว่านี้" [ 128 ] [ 123 ] [ 129 ]ความสนใจอย่างต่อเนื่องของออสเลอร์ในโรคฮันติงตัน ประกอบกับอิทธิพลของเขาในสาขาการแพทย์ ช่วยให้การรับรู้และความรู้เกี่ยวกับความผิดปกติแพร่กระจายไปทั่ววงการแพทย์อย่างรวดเร็ว[ 123 ]นักวิทยาศาสตร์ในยุโรปแสดงความสนใจอย่างมาก รวมถึงหลุยส์ เธโอฟิล โจเซฟ แลน ดูซี เดซิเร - มากลัวร์ บูร์เนวิลล์ คา มิ ลโล โกลจีและโจเซฟ จูลส์ เดอเจรีนและจนถึงสิ้นศตวรรษ งานวิจัยเกี่ยวกับโรคฮันติงตันส่วนใหญ่มีต้นกำเนิดมาจากยุโรป[ 123 ]เมื่อสิ้นสุดศตวรรษที่ 19 งานวิจัยและรายงานเกี่ยวกับโรคฮันติงตันได้รับการตีพิมพ์ในหลายประเทศ และโรคนี้ได้รับการยอมรับว่าเป็นภาวะที่เกิดขึ้นทั่วโลก[ 123 ]
ในช่วงการค้นพบการถ่ายทอดทางพันธุกรรมแบบเมนเดลอีกครั้งในช่วงต้นศตวรรษที่ 20 โรค HD ถูกนำมาใช้เป็นตัวอย่างของการถ่ายทอดทางพันธุกรรมแบบเด่นบนโครโมโซมร่างกาย[ 123 ]วิลเลียม เบตสัน นักชีววิทยาชาวอังกฤษใช้แผนผังลำดับวงศ์ตระกูลของครอบครัวที่ได้รับผลกระทบเพื่อยืนยันว่าโรค HD มีรูปแบบการถ่ายทอดทางพันธุกรรมแบบเด่นบนโครโมโซมร่างกาย[ 130 ] [ 125 ]รูปแบบการถ่ายทอดทางพันธุกรรมที่แข็งแกร่งนี้กระตุ้นให้นักวิจัยหลายคน รวมถึงสมิธ อีลี เจลลิฟฟ์พยายามติดตามและเชื่อมโยงสมาชิกในครอบครัวจากการศึกษาครั้งก่อนๆ[ 123 ]เจลลิฟฟ์รวบรวมข้อมูลจากทั่วรัฐนิวยอร์กและตีพิมพ์บทความหลายฉบับเกี่ยวกับลำดับวงศ์ตระกูลของโรค HD ในนิวอิงแลนด์ [ 131 ] งาน วิจัยของเจลลิฟฟ์กระตุ้นความสนใจของ ชาร์ลส์ เดเวนพอร์ตเพื่อนร่วมวิทยาลัยของเขาซึ่งมอบหมายให้เอลิซาเบธ มันซีย์ ทำการศึกษาภาคสนามครั้งแรกในชายฝั่งตะวันออกของสหรัฐอเมริกาเกี่ยวกับครอบครัวที่เป็นโรค HD และสร้างแผนผังลำดับวงศ์ตระกูลของพวกเขา[ 132 ]เดเวนพอร์ตใช้ข้อมูลนี้เพื่อบันทึกอายุที่เริ่มมีอาการและช่วงของอาการของ HD ที่แตกต่างกัน เขาอ้างว่ากรณีส่วนใหญ่ของ HD ในสหรัฐอเมริกาสามารถสืบย้อนกลับไปถึงบุคคลเพียงไม่กี่คนได้[ 132 ]งานวิจัยนี้ได้รับการเสริมแต่งเพิ่มเติมในปี 1932 โดยPR Vessieซึ่งทำให้แนวคิดที่ว่าพี่น้องสามคนที่ออกจากอังกฤษในปี 1630 มุ่งหน้าไปยังบอสตันเป็นบรรพบุรุษของ HD ในสหรัฐอเมริกา เป็นที่นิยม [ 133 ]การอ้างว่าบรรพบุรุษที่เก่าแก่ที่สุดได้รับการพิสูจน์แล้วและ อคติ ทางพันธุกรรมของงานของ Muncey, Davenport และ Vessie มีส่วนทำให้เกิดความเข้าใจผิดและอคติเกี่ยวกับ HD [ 125 ] Muncey และ Davenport ยังทำให้แนวคิดที่ว่าในอดีต บางคนที่เป็น HD อาจถูกคิดว่าถูกวิญญาณเข้าสิงหรือเป็นเหยื่อของเวทมนตร์และบางครั้งก็ถูกสังคมรังเกียจหรือเนรเทศ เป็นที่นิยม [ 134 ] [ 135 ]แนวคิดนี้ยังไม่ได้รับการพิสูจน์ผ่านการวิจัยทางวิทยาศาสตร์ นักวิจัยพบหลักฐานที่ขัดแย้งกัน ตัวอย่างเช่น ชุมชนของครอบครัวที่จอร์จ ฮันติงตันศึกษานั้นได้ให้การต้อนรับผู้ที่มีอาการของ HD อย่างเปิดเผย[ 125 ] [ 134 ]
การค้นหาสาเหตุของอาการนี้ได้รับการพัฒนาอย่างมากในปี พ.ศ. 2511 เมื่อมูลนิธิโรคทางพันธุกรรม (HDF) ก่อตั้งขึ้นโดยมิลตัน เวกซ์เลอร์นักจิตวิเคราะห์ที่อยู่ในลอสแอนเจลิส รัฐแคลิฟอร์เนียซึ่งภรรยาของเขา ลีโอโนเร ซาบิน ได้รับการวินิจฉัยว่าเป็นโรคฮันติงตันเมื่อต้นปีนั้น[ 136 ]พี่ชายทั้งสามคนของซาบินก็เป็นโรคนี้เช่นกัน
มูลนิธิมีส่วนร่วมในการสรรหานักวิทยาศาสตร์มากกว่า 100 คนในโครงการความร่วมมือโรคฮันติงตันระหว่างสหรัฐอเมริกาและเวเนซุเอลา ซึ่งทำงานในช่วงระยะเวลา 10 ปีตั้งแต่ปี 1979 เพื่อค้นหาสาเหตุทางพันธุกรรม[ 137 ]ซึ่งประสบความสำเร็จในปี 1983 เมื่อมีการระบุตำแหน่งยีนที่เป็นสาเหตุโดยประมาณ[ 116 ]และในปี 1993 ยีนดังกล่าวได้รับการระบุตำแหน่งอย่างแม่นยำที่โครโมโซม 4 (4p16.3) [ 138 ]การศึกษานี้มุ่งเน้นไปที่ประชากรในหมู่บ้านที่แยกตัวออกไปสองแห่งในเวเนซุเอลา ได้แก่ บาร์รังกิตัสและลากูเนตัส ซึ่งมีการแพร่ระบาดของโรคฮันติงตันสูงผิดปกติ และเกี่ยวข้องกับผู้คนมากกว่า 18,000 คน ส่วนใหญ่มาจากครอบครัวขยายเดียวกัน และส่ง ผลให้โรคฮันติงตันเป็นตำแหน่งโรคออโตโซม แรก ที่พบโดยใช้การวิเคราะห์การเชื่อมโยงทางพันธุกรรม[ 138 ] [ 139 ]ในบรรดานวัตกรรมอื่นๆ โครงการนี้ได้พัฒนา วิธี การทำเครื่องหมายดีเอ็นเอซึ่งเป็นขั้นตอนสำคัญที่ทำให้โครงการจีโนมมนุษย์เป็นไปได้[ 137 ]
ในขณะเดียวกัน ก็มีการค้นพบที่สำคัญเกี่ยวกับกลไกของความผิดปกติ รวมถึงการค้นพบโดย กลุ่มวิจัยของ Anita Hardingเกี่ยวกับผลกระทบของความยาวของยีน[ 140 ]
การจำลองโรคในสัตว์หลายชนิด เช่น หนู ทรานส์เจนิกที่พัฒนาขึ้นในปี 1996 ทำให้สามารถทำการทดลองขนาดใหญ่ขึ้นได้ เนื่องจากสัตว์เหล่านี้มีการเผาผลาญ ที่เร็วกว่า และมีอายุขัยสั้นกว่ามนุษย์มาก ผลการทดลองจึงได้รับเร็วขึ้น ซึ่งช่วยเร่งการวิจัย การค้นพบในปี 1997 ว่าชิ้นส่วน mHTT เกิดการพับตัวผิดปกตินำไปสู่การค้นพบการรวมตัวของนิวเคลียสที่เกิดจากชิ้นส่วนเหล่านั้น ความก้าวหน้าเหล่านี้ได้นำไปสู่การวิจัยที่กว้างขวางมากขึ้นเกี่ยวกับโปรตีนที่เกี่ยวข้องกับโรค การรักษาด้วยยาที่เป็นไปได้ วิธีการดูแลรักษา และตัวยีนเอง[ 123 ] [ 141 ]
เครือข่ายการดูแลและการสนับสนุนที่พัฒนาขึ้นในเวเนซุเอลาและโคลอมเบียในช่วงโครงการวิจัยในช่วงทศวรรษ 1970 ถึง 2000 ในที่สุดก็ถูกกัดเซาะโดยปัจจัยต่างๆ เช่นวิกฤตการณ์ที่กำลังดำเนินอยู่ในเวเนซุเอลาและการเสียชีวิตของนักวิจัยชั้นนำในโคลอมเบีย (Jorge Daza Barriga) [ 142 ]แพทย์กำลังพยายามฟื้นฟูเครือข่ายเหล่านี้ เพราะผู้คนที่ได้มีส่วนร่วมในวิทยาศาสตร์ของ HD โดยการเข้าร่วมในการศึกษาเหล่านี้สมควรได้รับการดูแลติดตามผลที่เพียงพอ สังคมอื่นๆ ในโลกที่ได้รับประโยชน์จากความก้าวหน้าทางวิทยาศาสตร์ที่เกิดขึ้นนั้น ควรจะตอบแทนผู้ที่เข้าร่วมในการวิจัยอย่างน้อยที่สุด[ 142 ]
เดิมทีอาการนี้เรียกว่าโรคฮันติงตัน (Huntington's chorea) แต่ต่อมาได้เปลี่ยนชื่อเป็นโรคฮันติงตัน (Huntington's disease) เนื่องจากไม่ใช่ผู้ป่วยทุกคนที่จะมีอาการชักกระตุก นอกจากนี้ ปัญหาด้านการรับรู้และพฤติกรรมยังเป็นส่วนสำคัญของอาการของโรคฮันติงตันอีกด้วย[ 143 ]
สังคมและวัฒนธรรม
จริยธรรม
การตรวจทางพันธุกรรมสำหรับโรคฮันติงตันได้ก่อให้เกิดประเด็นทางจริยธรรมหลายประการ ซึ่งรวมถึงการกำหนดว่าบุคคลควรมีวุฒิภาวะเท่าใดจึงจะถือว่ามีสิทธิ์ได้รับการตรวจ การรับรองความลับของผลการตรวจ และการตัดสินใจว่าบริษัทควรได้รับอนุญาตให้ใช้ผลการตรวจเพื่อประกอบการตัดสินใจเกี่ยวกับการจ้างงาน ประกันชีวิต หรือเรื่องทางการเงินอื่นๆ หรือไม่ มีข้อโต้แย้งเกิดขึ้นเมื่อชาร์ลส์ เดเวนพอร์ตเสนอในปี 1910 ว่าควรใช้การทำหมันแบบบังคับและ การควบคุม การเข้าเมือง สำหรับผู้ป่วยโรคบางชนิด รวมถึงโรคฮันติงตัน ซึ่งเป็นส่วนหนึ่งของ ขบวนการยูจีนิกส์[ 144 ]การปฏิสนธิในหลอดทดลองมีปัญหาบางประการเกี่ยวกับการใช้ตัวอ่อน การวิจัยโรคฮันติงตันบางส่วนมีปัญหาทางจริยธรรมเนื่องจากการใช้การทดลองในสัตว์และเซลล์ต้นกำเนิดจากตัวอ่อน[ 145 ] [ 146 ]
การพัฒนาการทดสอบวินิจฉัยโรค HD ที่แม่นยำได้ก่อให้เกิดความกังวลทางสังคม กฎหมาย และจริยธรรมเกี่ยวกับการเข้าถึงและการใช้ผลลัพธ์ของบุคคล[ 147 ] [ 148 ]แนวทางและขั้นตอนการทดสอบจำนวนมากมีขั้นตอนที่เข้มงวดสำหรับการเปิดเผยและการรักษาความลับ เพื่อให้บุคคลสามารถตัดสินใจได้ว่าจะรับผลลัพธ์เมื่อใดและอย่างไร และผลลัพธ์นั้นจะเปิดเผยให้ใครทราบ[ 29 ]บริษัทประกันภัยและธุรกิจต่างๆ ต้องเผชิญกับคำถามว่าควรใช้ผลการทดสอบทางพันธุกรรมเมื่อประเมินบุคคล เช่น สำหรับการประกันชีวิตหรือการจ้างงานหรือไม่ บริษัทประกันภัยของสหราชอาณาจักรตกลงกับกระทรวงสาธารณสุขและสังคมสงเคราะห์ว่าจนถึงปี 2017 ลูกค้าไม่จำเป็นต้องเปิดเผยการทดสอบทางพันธุกรรมเชิงพยากรณ์ให้พวกเขาทราบ แต่ข้อตกลงนี้ได้ยกเว้นการทดสอบโรคฮันติงตันที่ได้รับการอนุมัติจากรัฐบาลอย่างชัดเจนเมื่อออกกรมธรรม์ที่มีมูลค่าเกิน500,000ปอนด์[ 149 ] [ 150 ]เช่นเดียวกับภาวะทางพันธุกรรมที่รักษาไม่ได้อื่นๆ ที่เกิดขึ้นในภายหลัง การทดสอบก่อนมีอาการในเด็กหรือวัยรุ่นถือเป็นเรื่องที่น่าสงสัยทางจริยธรรม เนื่องจากจะไม่มีประโยชน์ทางการแพทย์สำหรับบุคคลนั้น มีฉันทามติในการทดสอบเฉพาะบุคคลที่ถือว่ามีวุฒิภาวะทางสติปัญญาแล้วเท่านั้น แม้ว่าจะมีการโต้แย้งว่าผู้ปกครองมีสิทธิ์ในการตัดสินใจแทนบุตรหลานของตนก็ตาม ด้วยการขาดการรักษาที่มีประสิทธิภาพ การทดสอบบุคคลที่อายุต่ำกว่าเกณฑ์ที่กฎหมายกำหนดและไม่ได้รับการตัดสินว่ามีความสามารถถือว่าผิดจริยธรรมในกรณีส่วนใหญ่[ 55 ] [ 151 ] [ 152 ]
มีข้อกังวลด้านจริยธรรมที่เกี่ยวข้องกับการตรวจทางพันธุกรรมก่อนคลอดหรือการวินิจฉัยทางพันธุกรรมก่อนการฝังตัวเพื่อให้แน่ใจว่าเด็กจะไม่เกิดมาพร้อมกับโรคที่กำหนด[ 153 ]ตัวอย่างเช่น การตรวจก่อนคลอดทำให้เกิดประเด็นเรื่องการทำแท้งแบบเลือก ซึ่งเป็นทางเลือกที่บางคนมองว่ายอมรับไม่ได้[ 153 ]เนื่องจากเป็นโรคที่ถ่ายทอดทางพันธุกรรมแบบเด่น จึงมีปัญหาในกรณีที่ผู้ปกครองไม่ต้องการทราบการวินิจฉัยของตนเอง ซึ่งจะทำให้ต้องเก็บขั้นตอนบางส่วนเป็นความลับจากผู้ปกครอง[ 153 ]
องค์กรสนับสนุน

ในปี พ.ศ. 2511 หลังจากพบโรค HD ในครอบครัวของภรรยา ดร.มิลตัน เว็กซ์เลอร์จึงได้รับแรงบันดาลใจให้ก่อตั้งมูลนิธิโรคทางพันธุกรรม (HDF) โดยมีเป้าหมายเพื่อรักษาโรคทางพันธุกรรมด้วยการประสานงานและสนับสนุนการวิจัย[ 19 ]มูลนิธิและแนนซี เว็กซ์ เลอร์ บุตรสาวของเว็กซ์เลอร์ เป็นส่วนสำคัญของทีมวิจัยในเวเนซุเอลาที่ค้นพบยีน HD [ 19 ]
ในเวลาเดียวกันกับการก่อตั้ง HDF มาร์จอรี กัทรีได้ช่วยก่อตั้งคณะกรรมการต่อต้านโรคฮันติงตัน (ปัจจุบันคือสมาคมโรคฮันติงตันแห่งอเมริกา ) หลังจากที่สามีของเธอ วู้ ดดี้ กัทรีนักร้องนักแต่งเพลง โฟล์ค เสียชีวิตจากภาวะแทรกซ้อนของโรคฮัน ติงตัน [ 20 ]
นับตั้งแต่นั้นมา องค์กรสนับสนุนและวิจัยได้ก่อตั้งขึ้นในหลายประเทศทั่วโลกและช่วยเพิ่มความตระหนักรู้ของสาธารณชนเกี่ยวกับโรคฮันติงตัน องค์กรเหล่านี้จำนวนหนึ่งร่วมมือกันในองค์กรหลัก เช่น สมาคมฮันติงตันนานาชาติและเครือข่ายโรคฮันติงตันแห่งยุโรป[ 154 ]องค์กรสนับสนุนหลายแห่งจัดงานสร้างความตระหนักรู้เกี่ยวกับโรคฮันติงตันเป็นประจำทุกปี ซึ่งบางงานได้รับการรับรองจากรัฐบาลของประเทศนั้นๆ ตัวอย่างเช่นวุฒิสภาสหรัฐอเมริกากำหนด ให้วันที่ 6 มิถุนายนเป็น "วันสร้างความตระหนักรู้เกี่ยวกับโรคฮันติงตันแห่งชาติ" [ 155 ]มีองค์กรมากมายที่ให้การสนับสนุนและให้ข้อมูลแก่ผู้ที่ได้รับผลกระทบจากโรคฮันติงตัน รวมถึงสมาคมโรคฮันติงตันในสหราชอาณาจักร ผู้ให้ทุนวิจัยรายใหญ่ที่สุดคือมูลนิธิริเริ่มการรักษาโรคฮันติงตัน (CHDI) [ 156 ]
ทิศทางการวิจัย
การวิจัยเกี่ยวกับกลไกของ HD มุ่งเน้นไปที่การระบุการทำงานของ Htt วิธีที่ mHtt แตกต่างหรือรบกวนการทำงานของ Htt และพยาธิสภาพของสมองที่โรคก่อให้เกิด[ 157 ]การวิจัยดำเนินการโดยใช้วิธีในหลอดทดลองสัตว์ดัดแปลงพันธุกรรม (เรียกอีกอย่างว่าแบบจำลองสัตว์ทรานส์เจนิก ) และอาสาสมัครมนุษย์ แบบจำลองสัตว์มีความสำคัญต่อการทำความเข้าใจกลไกพื้นฐานที่ก่อให้เกิดโรค และเพื่อสนับสนุนขั้นตอนเริ่มต้นของ การ พัฒนายา[ 141 ] การระบุยีนที่เป็นสาเหตุทำให้สามารถพัฒนาสิ่งมีชีวิตดัดแปลงพันธุกรรม ได้หลายชนิด รวมถึง หนอน ตัว กลม แมลงวันผลไม้Drosophila และสัตว์เลี้ยงลูกด้วยนมดัดแปลงพันธุกรรมได้แก่ หนู หนูแรต แกะ หมู และลิง ที่แสดงออกถึงฮันติงตินกลายพันธุ์และพัฒนาการเสื่อมของระบบประสาท อย่างต่อเนื่อง และอาการคล้าย HD [ 141 ]
มีการวิจัยโดยใช้วิธีการต่างๆ มากมายเพื่อป้องกัน HD หรือชะลอการลุกลามของ โรค [ 157 ]กลยุทธ์การปรับเปลี่ยนโรคสามารถแบ่งออกได้เป็น 3 ประเภทใหญ่ๆ ได้แก่ การลดระดับโปรตีนฮันติงตินกลายพันธุ์ (รวมถึงการตัดต่อยีนและการปิดกั้นยีน ) วิธีการที่มุ่งเป้าไปที่การปรับปรุงการอยู่รอดของเซลล์ประสาทโดยการลดอันตรายที่เกิดจากโปรตีนต่อเส้นทางและกลไกของเซลล์เฉพาะ (รวมถึงการรักษาสมดุลของโปรตีนและการยับยั้งฮิสโตนดีอะเซทิเลส ) และกลยุทธ์ในการทดแทนเซลล์ประสาทที่สูญเสียไป นอกจากนี้ ยังมีการพัฒนายาบำบัดใหม่ๆ เพื่อปรับปรุงการทำงานของสมอง ซึ่งมุ่งเน้นการรักษาตามอาการมากกว่าการปรับเปลี่ยนโรคและรวมถึงสารยับยั้งฟอสโฟไดเอสเตอเรส[ 158 ] [ 159 ]
มูลนิธิCHDIให้ทุนสนับสนุนโครงการวิจัยและตีพิมพ์ผลงานมากมาย[ 160 ]มูลนิธิ CHDI เป็นผู้ให้ทุนสนับสนุนการวิจัยโรคฮันติงตันที่ใหญ่ที่สุดในโลก และมีเป้าหมายที่จะค้นหาและพัฒนายาที่จะชะลอการลุกลามของโรคฮันติงตัน[ 156 ] [ 161 ]เดิมที CHDI เป็นที่รู้จักในชื่อมูลนิธิ High Q ในปี 2549 ได้ใช้เงิน 50 ล้านดอลลาร์สหรัฐในการวิจัยโรคฮันติงตัน[ 156 ] CHDI ร่วมมือกับห้องปฏิบัติการทางวิชาการและเชิงพาณิชย์มากมายทั่วโลก และมีส่วนร่วมในการกำกับดูแลและจัดการโครงการวิจัย ตลอดจนการให้ทุนสนับสนุน[ 162 ]
ลดการผลิตฮันติงติน
การปิดกั้นยีนมีเป้าหมายเพื่อลดการผลิตโปรตีนกลายพันธุ์ เนื่องจาก HD เกิดจากยีนเด่นเพียงยีนเดียวที่เข้ารหัสโปรตีนที่เป็นพิษ การทดลองปิดกั้นยีนในแบบจำลองหนูแสดงให้เห็นว่าเมื่อการแสดงออกของmHttลดลง อาการจะดีขึ้น[ 163 ]ความปลอดภัยของการแทรกแซง RNAและ วิธีการปิดกั้นยีน ด้วยโอลิโกนิวคลีโอไทด์เฉพาะอัลลีล (ASO) ได้รับการพิสูจน์แล้วในหนูและลิงมาคาก[ 164 ] [ 165 ]แนวทางการปิดกั้นเฉพาะอัลลีลพยายามที่จะกำหนดเป้าหมายmHTT ในขณะที่ปล่อยให้ HTTชนิดปกติไม่ได้รับผลกระทบโดยใช้ประโยชน์จากโพลีมอร์ฟิซึมที่มีอยู่ในอัลลีลกลายพันธุ์เท่านั้น[ 166 ]
การทดลองปิดการทำงานของยีนครั้งแรกในมนุษย์ที่เป็นโรค HD เริ่มขึ้นในปี 2015 โดยทดสอบความปลอดภัยของ IONIS-HTTRx ซึ่งผลิตโดยIonis PharmaceuticalsและนำโดยUCL Institute of Neurology [ 167 ] [ 168 ] ตรวจพบและวัดปริมาณโปรตีนฮันติงตินกลายพันธุ์เป็นครั้งแรกในน้ำไขสันหลัง ของผู้ที่มียีนกลายพันธุ์ HD ในปี 2015 โดยใช้การตรวจ วิเคราะห์ภูมิคุ้มกันแบบ "นับโมเลกุลเดี่ยว" แบบใหม่[ 169 ]ซึ่งเป็นวิธีโดยตรงในการประเมินว่าการรักษาที่ลดระดับฮันติงตินนั้นได้ผลตามที่ต้องการหรือไม่[ 170 ] [ 171 ] การทดลองระยะที่ 3 ของสารประกอบนี้ ซึ่งเปลี่ยนชื่อเป็น tominersen และได้รับการสนับสนุนโดยRoche Pharmaceuticalsเริ่มขึ้นในปี 2019 แต่ถูกระงับในปี 2021 หลังจากคณะกรรมการตรวจสอบความปลอดภัยสรุปว่าความสมดุลระหว่างความเสี่ยงและผลประโยชน์ไม่เอื้ออำนวย[ 172 ]การทดลองการบำบัดด้วยยีนที่ลดระดับฮันติงตินซึ่งดำเนินการโดยบริษัทเภสัชกรรมดัตช์uniQure Biopharmaเริ่มขึ้นในปี 2019 และมีการประกาศการทดลองสารประกอบโมดูเลเตอร์การตัดต่อยีนที่ลดระดับฮันติงตินแบบรับประทานหลายรายการ[ 173 ]ในจำนวนนี้votoplamผ่านการทดลองเฟส 2 ในปี 2025 [ 174 ] กำลังมีการศึกษาเทคนิค การตัดต่อยีนเพื่อพยายามซ่อมแซมจีโนมที่มียีนผิดพลาดซึ่งเป็นสาเหตุของ HD โดยใช้เครื่องมือต่างๆ เช่นCRISPR/Cas9 [ 159 ] PTC Therapeuticsกำลังประเมินโมเลกุลขนาดเล็กที่กระตุ้น การรวมเอ็ก ซอนพิษใน ทรานสคริปต์ HTTเป็นกลยุทธ์การรักษาเพื่อลดการแสดงออกของ HTT [ 175 ] [ 176 ]
ในปี 2025 นักวิจัยที่University College Londonรายงานผลเบื้องต้นจากAMT-130 ซึ่งเป็นการทดลองทางคลินิกระยะที่ 1/2 ของ uniQure เกี่ยวกับการบำบัดด้วยยีนเพื่อลดระดับฮันติงติน โดยแสดงให้เห็นว่าการดำเนินของโรคช้าลง 75% ในช่วงสามปี ข้อมูลการศึกษาฉบับเต็มยังไม่ได้รับการเผยแพร่หรือตีพิมพ์ อย่างเป็นทางการ [ 177 ] [ 178 ] [ 179 ] AMT-130 เป็นการบำบัดด้วยยีนโดยใช้ เวกเตอร์ ไวรัสอะเดโนแอสโซซิเอตชนิดที่ 5 (AAV5) ที่บรรจุไมโครอาร์เอ็นเอเทียมเพื่อปิดการทำงานของยีน ส่งผลให้การแสดงออกของโปรตีนฮันติงตินลดลง[ 180 ]การบำบัดนี้จะถูกส่งตรงไปยังโครงสร้างสมองส่วนลึกของนิวเคลียสคอเดตและพูตาเมนผ่านขั้นตอนการผ่าตัดระบบประสาทที่นำทางด้วย MRI [ 181 ] [ 182 ]
การกำจัดฮันติงตินที่เพิ่มขึ้น
กลยุทธ์อีกประการหนึ่งในการลดระดับของฮันติงตินกลายพันธุ์คือการเพิ่มอัตราที่เซลล์สามารถกำจัดมันได้[ 183 ]เนื่องจาก mHTT (และโปรตีนรวมกลุ่ม อื่นๆ อีกมากมาย ) อาจถูกย่อยสลายโดยออโตฟาจีการเพิ่มอัตราของออโตฟาจีจึงมีศักยภาพในการลดระดับของ mHTT และด้วยเหตุนี้จึงช่วยบรรเทาอาการของโรคได้[ 184 ]ตัวกระตุ้นออโตฟาจีทางเภสัชวิทยาและทางพันธุกรรมได้รับการทดสอบในแบบจำลอง HD หลายแบบ หลายชนิดแสดงให้เห็นว่าสามารถลดระดับ mHtt และลดความเป็นพิษในหนูได้[ 183 ]
การเพิ่มอัตราการอยู่รอดของเซลล์
แนวทางต่างๆ ที่มุ่งเป้าไปที่การปรับปรุงการอยู่รอดของเซลล์เมื่อมีฮันติงตินกลายพันธุ์ ได้แก่ การแก้ไขการควบคุมการถอดรหัสโดยใช้สารยับยั้งฮิสโตนดีอะเซ ทิเลส การปรับการรวมตัวของฮันติงติน การปรับปรุงการเผาผลาญและการทำงานของไมโตคอนเดรียและการฟื้นฟูการทำงานของไซแนปส์[ 163 ]
การทดแทนเซลล์ประสาท
การบำบัดด้วยสเต็มเซลล์ใช้เพื่อทดแทนเซลล์ประสาทที่เสียหายโดยการปลูกถ่ายสเต็มเซลล์เข้าไปในบริเวณสมองที่ได้รับผลกระทบ การทดลองในหนูและหนูทดลองที่เป็นแบบจำลองของโรค HD ได้ผลลัพธ์ที่เป็นบวก[ 185 ]สเต็มเซลล์ยังถูกนำมาใช้เพื่อศึกษาโรค HD ในห้องปฏิบัติการอีกด้วย[ 186 ]
เฟอร์โรพโทซิส
เฟอร์โรพโทซิสเป็นรูปแบบหนึ่งของการตายของเซลล์ที่มีการควบคุม โดยมีลักษณะเฉพาะคือการสะสมของไฮโดรเปอร์ออกไซด์ของไขมันที่ขึ้นอยู่กับธาตุเหล็กจนถึงระดับที่เป็นอันตราย ถึงชีวิต เฟอร์โรพโทซิสที่เกิดจาก ALOX5ทำหน้าที่เป็นเส้นทางการตายของเซลล์เมื่อเกิดความเครียดจากออกซิเดชันใน HD [ 187 ]สารยับยั้งเฟอร์โรพโทซิสมีฤทธิ์ป้องกันในแบบจำลองของความผิดปกติของสมองที่เสื่อมลง รวมถึงโรคพาร์กินสัน โรคฮันติงตัน และโรคอัลไซเมอร์[ 187 ]
การทดลองทางคลินิก
ในปี 2020 มีการทดลองทางคลินิก 197 รายการที่เกี่ยวข้องกับการบำบัดและไบโอมาร์กเกอร์ต่างๆ สำหรับ HD ซึ่งระบุว่าอยู่ระหว่างดำเนินการ กำลังรับสมัคร หรือเพิ่งเสร็จสิ้น[ 188 ]สารประกอบที่ทดลองแล้วแต่ไม่สามารถป้องกันหรือชะลอการลุกลามของ HD ได้แก่remacemide , coenzyme Q , riluzole , creatine , minocycline , ethyl- EPA , phenylbutyrateและdimebon [ 189 ]
ดูเพิ่มเติม
ลิงก์ภายนอก
 คำจำกัดความจากวิกิพีเดีย
คำจำกัดความจากวิกิพีเดีย สื่อจากคอมมอนส์
สื่อจากคอมมอนส์ ข้อความจาก Wikisource
ข้อความจาก Wikisource ตำราเรียนจากวิกิบุ๊ก
ตำราเรียนจากวิกิบุ๊ก แหล่งข้อมูลจาก Wikiversity
แหล่งข้อมูลจาก Wikiversity
- โครงการ HOPES ถูกเก็บถาวรเมื่อวันที่ 27 สิงหาคม 2020 ที่Wayback Machine – โครงการข้อมูล HD ของมหาวิทยาลัยสแตนฟอร์ด
- HDBuzz – ข่าวสารงานวิจัยด้าน HD ที่เขียนโดยนักวิทยาศาสตร์ด้วยภาษาที่เข้าใจง่าย